
Abstract
CHARGE syndrome is an autosomal dominant multisystem disorder caused by mutation in the CHD7 gene, encoding chromodomain helicase DNA-binding protein 7. Molecular diagnostic testing for CHD7 mutation has been available in a clinical setting since 2005. We report here the results from the first 642 unrelated proband samples submitted for testing. Thirty-two percent (n = 203) of patient samples had a heterozygous pathogenic variant identified. The lower mutation rate than that published for well-characterized clinical samples is likely due to referral bias, as samples submitted for clinical testing may be for “rule-out” diagnoses, rather than solely to confirm clinical suspicion. We identified 159 unique pathogenic mutations, and of these, 134 mutations were each seen in a single individual and 25 mutations were found in two to five individuals (n = 69). Of the 203 mutations, only 9 were missense, with 107 nonsense, 69 frameshift, and 15 splice-site mutations likely leading to haploinsufficiency at the cellular level. An additional 72 variations identified in the 642 tested samples (11%) were considered to have unknown clinical significance. Copy number changes (deletion/duplication of the entire gene or one/several exons) were found to account for a very small number of cases (n = 3). This cohort represents the largest CHARGE syndrome sample size to date and is intended to serve as a resource for clinicians, genetic counselors, researchers, and other diagnostic laboratories.
Introduction
CHARGE
De novo mutations in the gene encoding chromodomain helicase DNA-binding protein 7 (CHD7) are the major cause of CHARGE syndrome (Vissers et al., 2004). DNA sequencing detects CHD7 mutations in ∼58%-64% of patients clinically diagnosed with CHARGE syndrome (Vissers et al., 2004; Jongmans et al., 2006; Lalani et al., 2006). Of the CHD7 mutations reported thus far, ∼70% are nonsense or frameshift, 6%-13% are missense, and 7%-15% are splice site mutations (Vissers et al., 2004; Felix et al., 2006; Jongmans et al., 2006; Lalani et al., 2006; Sanlaville et al., 2006; Aramaki et al., 2007; Vuorela et al., 2007; Asakura et al., 2008; Bergman et al., 2008; Gennery et al., 2008; Wincent et al., 2008). Partial and whole gene deletions or duplications are rare, accounting for 3%-4% of pathogenic CHD7 mutations (Aramaki et al., 2006; Vuorela et al., 2007; Bergman et al., 2008; Wincent et al., 2008). Although germline transmission of CHD7 mutation has been reported (Pauli et al., 2009), the majority of mutations arise de novo.
GeneDx is a Clinical Laboratory Improvement Amendments-certified commercial laboratory that specializes in genetic testing for over 250 rare genetic disorders. Clinical testing of the CHD7 gene at GeneDx has been available since 2005 and is performed on patient specimens using sequence analysis and, when indicated or desired, copy number analysis. The clinical utility of CHD7 gene analysis is to confirm a clinical diagnosis of CHARGE syndrome or to resolve a differential diagnosis that may include diseases with similar or overlapping clinical presentations, such as Kallmann syndrome, 22q11 deletion syndrome, VACTERL association, and retinoic embryopathy. In addition, patients presenting with one or two of the clinical features of CHARGE syndrome, such as coloboma or choanal atresia, may also be referred for CHD7 testing as these patients could have an unusual presentation of the disease.
Here we present the results of 642 unrelated patient samples submitted to GeneDx for CHD7 mutation analysis. Based on recommendations by the American College of Medical Genetics (Richards et al., 2008) for designating a variant as pathogenic, 203 of the 642 samples tested (32%) contained a variant in the CHD7 gene that was considered pathogenic. This differs from other reports in the literature of 58%-64% positive rate of CHD7 sequencing in CHARGE patients, reflecting the frequent lack of detailed clinical data provided with samples submitted to a clinical service laboratory. Twenty five of the 159 unique mutations (16%) were observed more than once, suggesting the presence of mutational hotspots within CHD7. We could not determine the clinical significance of additional 72 variants because of lack of available parental samples, clinical information, or functional data.
Methods
Criteria for diagnosis are defined by physicians and genetic counselors and are not provided to us. Genomic DNA was purified from buccal swabs or peripheral blood lymphocytes by standard methods. The protein-encoding exons of the CHD7 gene, exons 2-38, were amplified using oligonucleotide primers targeting intronic sequence flanking CHD7 exons under standard polymerase chain reaction conditions and sequenced bidirectionally by capillary sequencing on an ABI3730, using primers designed and optimized by the clinical laboratory. To help in identifying polymorphisms, synonymous and nonsynonymous variants were examined for conservation with the zebrafish CHD7-like protein (XP_697956). The zebrafish sequence was chosen for this purpose because it aligns well with the human sequence, yet it has more divergence from human CHD7 than other available sequences. Human and mouse CHD7 proteins are 97.1% identical, and human and chicken have 91.9% identity, whereas human and zebrafish CHD7s have 64.2% identity.
Testing for a CHD7 exon deletion or duplication is now performed by the laboratory upon request, using either multiplex ligation-dependent probe amplification (MLPA) (SALSA MLPA kit P201-B1; MRC-Holland) or exon-level resolution oligonucleotide array comparative genomic hybridization (ExonArray). A proprietary CopyDx quantitative polymerase chain reaction method is used to confirm whole or partial gene deletions or duplications. We tested 11 samples by MLPA, 8 by exon array, and 4 by CopyDx after DNA sequencing showed no obvious disease-causing mutation and few or no heterozygous polymorphisms.
Results
Six hundred forty-two patients were referred by physicians and other authorized providers to GeneDx for clinical genetic testing of the CHD7 gene. For each specimen, the entire protein coding sequence of the CHD7 gene, along with intron sequence flanking each exon, was analyzed by DNA sequencing. Variants predicted to introduce premature stop codons or cause frameshifts were considered pathogenic. Variants involving the canonical splice donor-acceptor pair (GT-AG) were also considered pathogenic, in keeping with American College of Medical Genetics guidelines (Richards et al., 2008). Missense changes or other putative splicing changes were considered pathogenic if proven de novo by testing both parents for its absence, or if the change was identical to a previously reported de novo disease-associated mutation. Heterozygous polymorphisms were recorded to document the presence of two alleles.
We identified pathogenic CHD7 mutations in 203 (32%) patient samples. Two hundred of 203 mutations involved small, mostly single base changes, which were detected by DNA sequencing. These include 107 nonsense mutations, 69 frameshift mutations, 15 splicing mutations, and 9 missense mutations (Table 1 and Table 2). One nonsense mutation was identified in the blood sample from a parent who was presumed to be mosaic. Two of the nine mutations classified as missense are located in the last bases of exons 8 and 17. These two mutations could affect splicing, as mutations at the end of exons are reported to inhibit the ability of the exon to be recognized by splicing factors (Talerico and Berget, 1990). We observed 25 different mutations more than once, including 7 not previously reported (Vissers et al., 2004; Felix et al., 2006; Jongmans et al., 2006; Lalani et al., 2006; Writzl et al., 2007; Gennery et al., 2008) (Table 2). Of these 25 mutations, 20 are nonsense, 3 are frameshift, and 2 are missense. The mutations we have identified are distributed throughout the coding region and do not appear to be preferentially located within regions corresponding to functional domains. The locations of the single-base mutations in the CHD7 gene and corresponding protein are shown in Figure 1.
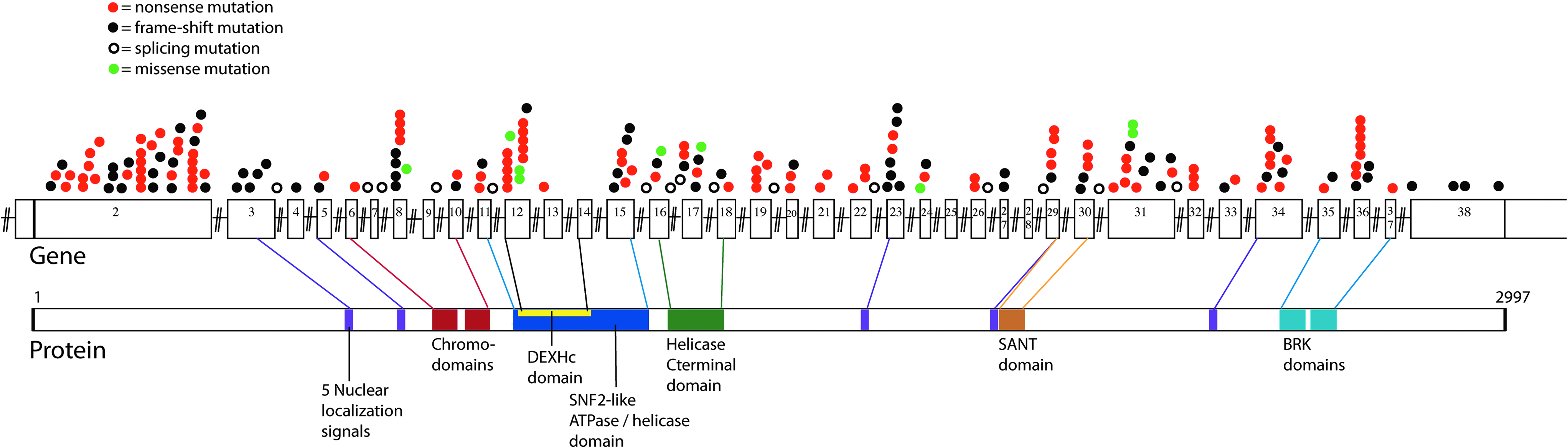
The CHD7 gene (top) and protein (bottom). Colored circles above exons 2-38 depict the location of 200 CHARGE-causing mutations. Overlapping circles indicate identical mutations. Protein domains are labeled and lines indicate where each protein domain is encoded on the gene. All DNA mutations that introduced stop codons or frameshifts were considered disease causing, as were mutations of the canonical splice donor-acceptor pair (GT-AG). Missense changes or other putative splicing changes were not considered disease causing unless the change was a de novo mutation not found in either parent or if it was reported in the literature as a de novo mutation.
Mutated bases in the human CHD7 cDNA were numbered based on accession number NM_017780. Gene and protein nomenclature follows recommendations (den Dunnen and Antonarakis, 2001). Bold font indicates recurrent mutations.
Conserved in zebrafish
CHD7 protein.
1Vissers et al. (2004).
2Aramaki et al. (2006).
3Jongmans et al. (2006).
4Lalani et al. (2006).
5Sanlaville et al. (2006).
6Felix et al. (2006).
7Delahaye et al. (2007).
8Vuorela et al. (2007).
9Writzl et al. (2007).
10Gennery et al. (2008).
11Wincent et al. (2008).
12Lee et al. (2009).
x = premature stop codon; f = frameshift mutation; s = splice site mutation; m = missense mutation; del = deletion; dup = duplication; ins = insertion.
Twenty-three specimens were further analyzed for copy number changes, using MLPA, CopyDx, or exon-level CGHarray. Of these cases, one duplication of exon 3, one whole gene deletion, and one deletion of exon 2 were identified.
A number of variants were identified in patient specimens that were novel or eluded classification (Table 3a). In most cases (n = 54) this is due to the lack of available parent samples. Such results are reported as being variants of unknown significance in patient reports. In another 18 cases, where we were able to test one or both parents, the variant was observed in a parent. However, without further information, these missense changes are difficult to classify. Germline transmission has been reported and may be due to germline mosaicism, somatic mosaicism, or inheritance of the mutation from a mildly affected parent. When inheritance of a novel missense change is observed in a molecular diagnostic setting, it is difficult to know if the parent is mosaic for a pathogenic mutation or if the variant is a benign polymorphism (Zlotogora, 1998). In these cases, clinical evaluation of the parent is recommended to the referring physician.
The inherited/de novo status of most is unknown unless otherwise indicated.
Conservation with zebrafish CHD7 protein is noted for coding mutations. Conservation with zebrafish CHD7 DNA sequence is noted for possible splice variations.
1Vissers et al. (2004).
2Aramaki et al. (2006).
3Jongmans et al. (2006).
4Lalani et al. (2006).
5Sanlaville et al. (2006).
6Felix et al. (2006).
7Delahaye et al. (2007).
8Vuorela et al. (2007).
9Writzl et al. (2007).
10Gennery et al. (2008).
11Wincent et al. (2008).
12Lee et al. (2009).
14Holak et al. (2008).
Blue-shaded rows: five mutations are 1-2 bp from an intron and we believe these to be splicing mutations. The IVS25-7G>A mutation, in five additional samples, has been previously reported but has not yet been identified as a de novo mutation.
Blues boxes: algorithm-predicted splice site mutations.
Green boxes: missense mutations.
Gray boxes: deletion, duplication, insertion mutations.
Bold font indicates recurrent mutations.
Twenty-two variants that may affect splicing were identified based on prediction algorithms such as SIFT (Lowe, 2004) and PolyPhen (Ramensky et al., 2002). Two of these were synonymous changes that could affect splicing, as has been observed in other diseases (Eriksson et al., 2003). Table 3a includes nonsynonymous missense variants that are presumed to be very rare, as they have been observed only once or twice in the 1284 alleles we have tested and were not found in the SNP databases. Without further information, we cannot determine the pathogenicity of these variants.
Table 3b lists variants characterized as benign polymorphisms. Twenty-nine polymorphisms were found in multiple individuals in this report or in previous reports, two were found in individuals also carrying disease-causing mutations, and one was homozygous.
A total of 370 specimens had no detectible mutations by sequencing, and we were able to test only 23 specimens for exonic copy number changes. It is not unusual to observe several polymorphisms in the CHD7 gene, and the observation of heterozygous positions ensures the presence of both alleles. In our negative samples, 76 altogether lacked heterozygous polymorphisms and another 88 specimens had only one to two polymorphisms. Although copy number changes are not common in the CHD7 gene, these samples (26% of all samples submitted) may be good candidates for deletion and duplication testing using other methods for detection.
Discussion
This report serves as a summary of the findings of CHD7 mutation analysis observed by one clinical diagnostic laboratory. Unlike other publications, we have not performed clinical evaluations on the patients in whom the analyses were performed, and these data must be regarded with that in mind.
We detected CHD7 mutations in 203 of 642 (∼32%) patient specimens referred to GeneDx for clinical testing. One hundred twenty of the 203 CHD7 mutations have not been previously reported. Consistent with previous reports, most of the mutations we detected are nonsense (n = 107; 52.7%) and frameshift (n = 69; 34%) mutations and are predicted to cause loss of function. Splicing (n = 15; 7.4%), missense (n = 9; 4.4%), and copy number changes (n = 3; 1.5%) are less common. There is a higher percentage of stop codons in our cohort than in the published literature (52.7% vs. 35.4%). Of 189 published mutations, there were 35.4% stop mutations, 33.3% frameshifts, 7%-15% splicing mutations, 6%-13% missense mutations, and 3% large deletion/duplications (Vissers et al., 2004; Felix et al., 2006; Jongmans et al., 2006; Lalani et al., 2006; Sanlaville et al., 2006; Aramaki et al., 2007; Vuorela et al., 2007; Asakura et al., 2008; Bergman et al., 2008; Gennery et al., 2008; Wincent et al., 2008).
Variants found in 11% (n = 72) of our 642 patient samples could not be classified as either pathogenic or benign, which clearly underscores the need for a functional assay, or at least the availability of parental samples for follow-up. The observation of mutations in 32% of the specimens evaluated is far lower than the 58%-64% reported by other groups. This reflects the variability in clinicians' use of molecular diagnostic testing, including the fact that many clinicians are considering a number of diagnoses in the differential when faced with a child who has some findings indicative of CHARGE syndrome.
Notably, there are 25 different mutations that have been observed more than once in our cohort. Six of these recurrent mutations were observed in four or more patient specimens. Each of these is a nonsense change involving a CGA arginine codon (R312X, R494X, R858X, R987X, R1036X, R2631X). This is consistent with reports that the CG dinucleotide is hypermutable to TG (Youssoufian et al., 1988; Antonarakis et al., 2000), making the arginine CGA codon uniquely vulnerable to transition to a nonsense mutation. Human CHD7 contains 27 arginine CGA codons. Despite these mutation hot spots, this should not influence how CHD7 mutation analysis is performed, given that overall these account for only a fraction (14.2%) of the total observed mutations.
In our study, we tested the DNA samples of both parents for the presence of a variant identified in their child in 25 families. This included testing for 12 nonsense, 6 frameshift, 6 missense, and 1 splice site mutation. Of these, only one mutation was identified in a parent and all other mutations had arisen de novo. Mosaicism for a p.E2546X nonsense mutation was observed in this individual's specimen. Sixteen cases of germline transmission of CHD7 mutation have been reported (Jongmans et al., 2006; Lalani et al., 2006; Delahaye et al., 2007; Jongmans et al., 2008; Vuorela et al., 2008; Wincent et al., 2008; Pauli et al., 2009). Some of these cases involve an affected or mildly affected parent, whereas in other cases the carrier parent is reported as unaffected. Two affected siblings have been reported in a family where the father had no detectable CHD7 gene mutation in lymphocyte DNA, but showed a mutation in ¼ of his sperm (Pauli et al., 2009).
The CHD7 gene is large at over 188 kb. Most laboratories seek mutations in the ∼9 kb that constitute the protein coding sequence and intron-exon junctions. As with most diagnostic tests, mutation analysis does not include the promoter, noncoding exons, or introns. Future full-gene sequencing using “next-generation” methods may increase the clinical sensitivity of diagnostic testing of CHARGE syndrome, revealing mutation deep in introns and promoter regions in other patients who carry a clinical diagnosis but are mutation negative using current methods.
Footnotes
Acknowledgments
P.C.S. is supported by grants from the National Institute of Child Health and Development (R01HD056369) and the National Human Genome Research Institute (R01HG004722).
Disclosure Statement
C.S. is currently an employee of Medco Health Solutions, Inc. Her contributions to this document are not to be construed as reflecting the views of Medco Health Solutions, Inc.