
Abstract
Aims: The connexin 26 coding gene (GJB2) is the primary causative gene for nonsyndromic sensorineural hearing impairment (NSSHI). More than 100 mutations in this gene have been reported to be linked to hearing impairment (HI), from mild to profound hearing loss. To precisely estimate the impact of GJB2 mutations in the Chinese population, a cross-sectional study was performed to analyze the auditory data of Chinese patients with NSSHI.Results: Two hundred ninety-five unrelated patients with NSSHI with biallelic mutations in GJB2 were recruited from seven provinces in Northern China from 2004 to 2008. The levels of HI and average pure tone audiometry were compared across different genotypes by χ2 testing. The subjects with the genotypes of combined truncating mutations had more cases of severe HI than the subjects with a genotype of several nontruncating mutations. It was also revealed that subjects carrying either c.[79G>A; 341A>G]+[79G>A; 341A>G] or c.[109G>A]+[79G>A; 341A>G] had significantly fewer cases of severe HI than the reference group of homozygous c.235delC, whereas the subjects carrying c.[235delC]+[176_191del16] had more cases of severe HI than the homozygous c.235delC group.Conclusions: This is the first study to clarify the correlations between different GJB2 biallelic genotypes and NSSHI phenotype in the Chinese population. The Chinese subjects with two truncating mutations in GJB2 were shown to correlate with more severe HI.
Introduction
Some mutations were verified to be pathogenic by functional studies in vitro, although the resulting phenotypes were mild HI, which included p.Val37Ile, p.Met34Thr, p.Leu90Pro, p.Arg184Pro, p.Trp77Arg, c.[-23+1G>A], and p.Arg127His. Nevertheless, some mutations were thought to be polymorphisms of high allelic frequency in carriers, such as [p.Val27Ile;p.Glu114Gly], but some functional studies found that calcium conductance in the cells transfected by plasmids carrying this mutation was damaged because of dysfunction of the gap junction protein (Bruzzone et al., 2003; Pilipenko et al., 2007). Despite these contradictory results, the study of c.250G>C (p.Val84Leu) (Beltramello et al., 2005) threw a light on new ways to explore functions of the gap junction protein, which revealed a key function of inositol triphosphate (IP3) in deafness etiology.
For the crucial role of GJB2 in etiology of NSSHI, several studies have been performed to reveal the correlation between genotypes and the phenotype clinically. It was clarified that combination of biallelic truncating mutations (frameshift, nonsense, insertion, and deletion) would lead to more severe hearing loss than those of two nontruncating mutations (missense, deletion, or insertion more than 3 times' nucleotides) in combination in the Caucasian and Japanese populations (Martini and Mazzoli, 1999; Oguchi et al., 2005; Yuan et al., 2007). However, this hypothesis has not yet been verified, although it is known that the Chinese population has the distinct spectrum of GJB2 mutations compared with other populations. Therefore, we enrolled and studied 295 Chinese deaf subjects carrying GJB2 biallelic mutations to evaluate the correlations between genotype and phenotype. These results would help precisely estimate the impact of GJB2 mutations in the Chinese population.
Materials and Methods
Subject recruitment
Subjects with NSSHI were sequentially recruited from our clinic from 2004 to 2008. Individuals with syndromic, unilateral, acquired, or dominant types of HI were excluded from this study. The clinical information comprised a complete history, physical examinations, and the audiometric examinations. Some also received radiological examinations to exclude dysplasia.
A total of 307 subjects with NSSHI who were screened with biallelic GJB2 mutations came from 7 provinces in Northern China, of whom 12 subjects were excluded because their audiometric data were incomplete, such as only data from the auditory brainstem response (ABR) test were available, which did not provide complete frequency-specific thresholds. To avoid potential biases, no familial subjects with identical genotypes were enrolled. Finally, detailed audiometric data from 295 persons were analyzed in this study. Information consent, blood samples, and clinical evaluations were obtained from all the participants according to the protocols approved by the review boards of the ethics committees of the Chinese People's Liberation Army General Hospital.
Audiological analysis
All subjects in this study were tested by pure tone audiometry and acoustic impedance admittance measurements in a sound proof room. Two hundred seventy subjects also received auditory brainstem response (ABR), and 65 received distortion product otoacoustic emissions (DPOAE). Sixty-four subjects who did not respond well to ABR also received auditory steady-state response (ASSR) tests. According to the pure tone averages (PTA) at frequencies from 0.5 kHz to 4 kHz (PTA0.5-4 kHz), average thresholds in the range of 21-40 dB were defined as “mild HI,” 41-70 dB as “moderate HI,” 71-95 dB as “severe HI,” and>95 dB as “profound HI”(Martini and Mazzoli, 1999).
GJB2 mutations analysis
All subjects were subjected to DNA sequencing of two exons in the GJB2 gene. However, two loci were not examined, which included c.[del(GJB6-D13S1830)], because it was seldom present in Chinese population (Yuan et al., 2007), and c.[-23+1G>A]. The causative mutations listed on the Connexin-Deafness HomePage and several controversial variants (e.g., c.109G>A (p.Val37Ile) and c.[79G>A; 341A>G]([p.Val27Ile; p.Glu114Gly]) were checked. In addition, some novel mutations with unclear functions were also screened, including missense mutations and insert mutations. Based on their functional implications (i.e., translated proteins), these loci were classified as nontruncating or truncating mutations. Missense mutations and deletions of 3 bp that resulted in a deletion of an amino acid were defined as nontruncating (NT) type. Similarly, splice site mutations, insertions, nonsense mutations, duplications, and deletions of more than 3 bp were defined as truncating mutations (T).
Statistical analysis
The linear regression of PTA0.5-4 kHz on age was performed to find out whether progression exists in a major genotype. Pearson's χ2 testing using the HI classification of mild, moderate, severe, and profound was performed to analyze whether the frequency distribution of HI was randomized in different genotype categories (T+T, T+NT, and NT+NT). Then, Fisher's exact probability testing with 2×2 contingency tables of appropriately dichotomized data was performed to find out the most frequent class of HI in each category. Here, PTA0.5-4 kHz as the grouping variable, the distribution frequency of each genotype was compared with the homozygous c.235delC group by Fisher's exact probability testing, which was applied with 2×2 contingency tables of appropriately dichotomized data at the median level (75th percentile [P75], 50th percentile [P50], 25th percentile [P25], 5th percentile [P5]) of the reference group.
Result
Sample information
Totally, 295 subjects (107 women and 188 men) with NSSHI were found to have two mutations in the GJB2 gene. Ages ranged from 5 months to 60 years, with the median age of 14, and the majority was within the age range of 2-21 years. Most of the subjects were of Han ethnicity, and some were minority groups, comprising of four Uighurs (1.3%), two Mongolians (0.7%), one Tibetan (0.3%), and two Hui (0.7%). Since very few minority were included, we did not separate the analysis by race. The difference of PTA0.5-4 kHz between two ears was from 0 to 27dB in the sample and mostly less than 15 dB.
Spectrum of mutations
The most frequent loci included six frameshift mutations c.235delC, c.[299_300delAT], c.[30_35delG], c.[30_35insG], c.[176_191del16], and c.[504_505insAAGG] and three missense mutations c.[79G>A;341A>G] ([p.Val27Ile;p.Glu114Gly]) and c.109G>A(p.Val37Ile). The remaining were several mutations with low frequency, including a nonsense mutation c.9G>A(p.Trp3X), three missense mutations c.257C>G (p.Thr86Arg), c.21G>A (p.Gln7Gln), c.427C>T (p.Arg143Trp), and a frameshift mutation c.[559_604dup46].
Spectrum of genotypes
There were totally 19 different genotypes, of which 6 were homozygous and 13 were compound heterozygous. Eleven genotypes were found in more than two subjects (Fig. 1), and the other 8 genotypes appeared in only one subject (Table 1). In all patients with biallelic mutations, 131 (45.9%) patients had homozygous c.235delC, and the allele frequency of c.235delC was 61.9% (365/590), similar to the ratio in the Japanese (Tsukada et al., 2010). However, a study on Chinese patients showed that frequency of c.235delC carrier in patients and in control group was 22.5% and 1%, respectively (Shi et al., 2004).

A scatter diagram of the PTA0.5-4 kHz of groups with specific genotypes. “n” indicates the number of each specific genotype group. “P50” indicates the median PTA0.5-4KhZ of each group. p-values were calculated using χ2 test by comparing each specific group to c.235delC homozygotes group as the reference. The threshold data were dichotomized at 108dB (P75 of the reference group), 100dB (P50), 86dB (P25), and 66dB (P5). PTA, pure tone average.
The c.257C>G (p. Thr86Arg) mutant protein did not form gap junctions. [11] c.9G>A(p.Trp3X) was a nonsense mutation, and c.21G>A(p.Gln7Gln) was a missense mutation. The pathogenic effect of the latter mutations remained unclear.
Fisher exact probability test was applied, and P values were calculated by comparing the median PTA0.5-4 kHz of each genotype with the median PTA0.5-4 kHz (108dB[∼P75 of the reference group], 100dB[∼P50], 86dB[∼P25], and 66dB[∼P5] of the reference group).
Results of DPOAE examination
Sixty-four sporadic subjects were screened with DPOAE. Fifty-four had uncertain conclusions because of their severe hearing loss beyond the upper limited threshold for DPOAE. The DPOAE was not recordable in the other ten subjects with PTA0.5-4 kHz less than 60 dB, in whom homozygous c.235delC were present in six subjects, c.[235delC]+[299_300delAT] in three, and c.[235delC]+[176_191del16] in one.
Onset age of HI in specific genotypes
The onset age was available in 199 subjects, ranging from 0 to 7 years, mainly during the 0-3 years, that is, prelingual. The newborns who did not pass the hearing screening were defined as onset on 0 year old. There were 11 subjects with postlingual deafness in this study, and their ages of onset ranged from 4 to 7 years. The genotypes represented in postlingual deafness included c.[235delC]+[235delC] which were present in four subjects, c.[235delC]+[299_300delAT] in three, c.[109G>A]+[79G>A; 341A>G] in one, c.[79G>A; 341A>G]+[79G>A; 341A>G] in two, and c.[30_35delG]+[299_300delAT] in one.
Relationship between age growth and HI
No linear regression was found between the ages and the PTA0.5-4 kHz in each specific genotype. So, no progression of HI was found in this sample while aging.
Distribution of HI among different genotype categories
Data from 287 subjects were shown in Figure 2. Only genotypes of the loci that were present in more than two subjects were analyzed. Per the nature of the mutations, these genotypes were further grouped into two categories, T+T and NT+NT. As shown in Figure 2, HI values in genotype categories were randomly distributed (χ2=3.39, p=0.183). Then, the 2×4 contingency (2 meant genotype category, and 4 meant HI levels) tables were reduced into 2×2 contingency tables to find the distribution frequency of each HI level in each category, and T+T category was found to have more cases of profound HI than the NT+NT category (p=0.074).
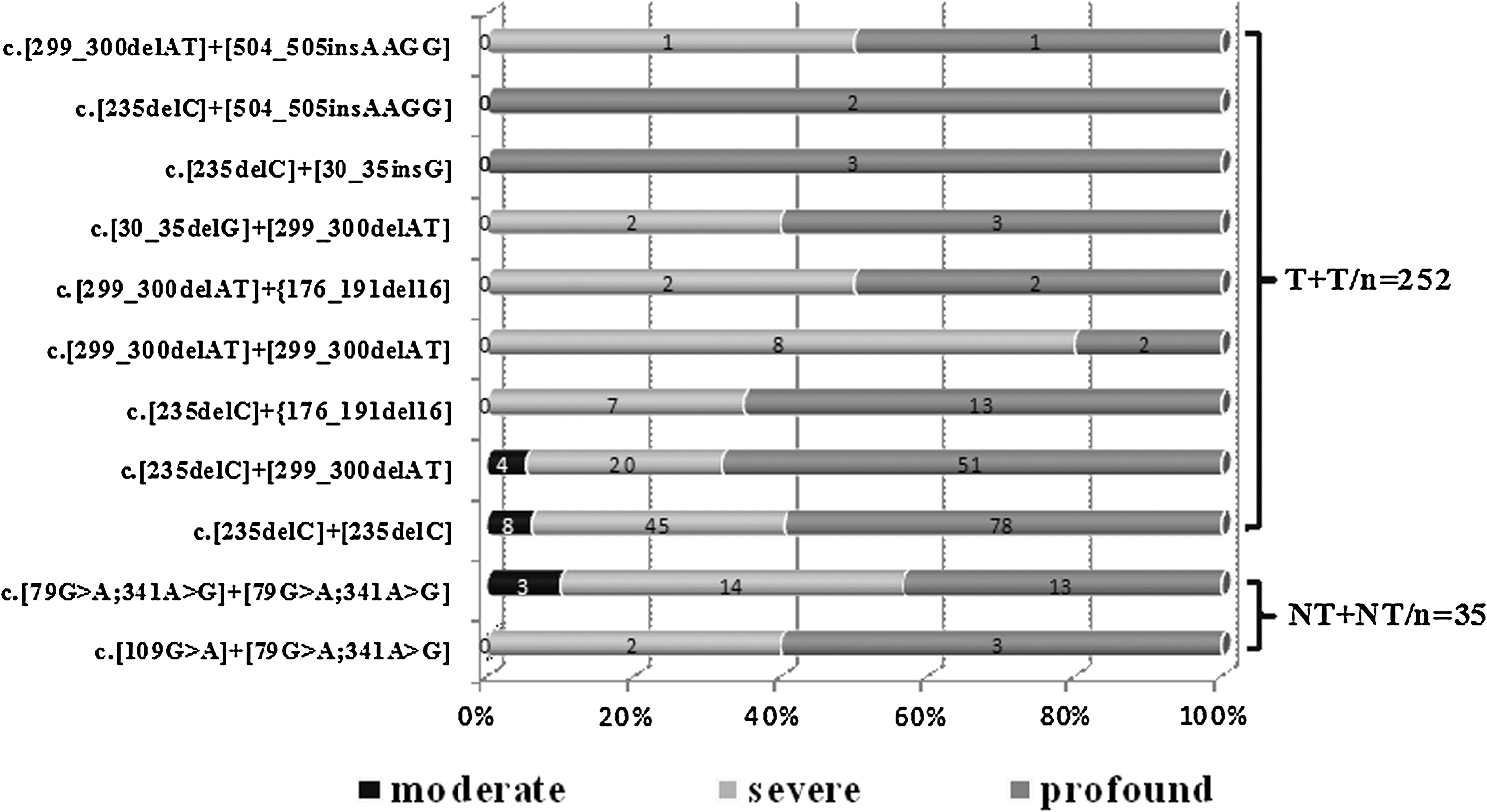
Distribution of the three classes of HI with specific genotype that represented in more than two subjects. The genotypes were divided into two categories, which were nontruncating+nontruncating (NT+NT) and truncating+truncating (T+T). The number of patients in each class and the genotypes are indicated under x-axis. The percentile of cases with different HI levels in each genotype was showed according to y-axis. χ2 test indicated that a nonrandom association existed between the degree of HI and the genotype category. HI, hearing impairment.
Comparison of HI between other GJB2 genotypes and homozygous c.235delC
First, we delineated the distribution of PTA of 287 subjects in 11 genotypic groups found in more than two subjects (Fig. 1). Then, the P50 PTA for each group was compared with the reference group (homozygous c.235delC). Though NT+NT category was not significantly related with the less HI comparing with the T+T category, there were two genotypes (c.[79G>A; 341A>G]+[79G>A; 341A>G], c.[109G>A]+[79G>A; 341A>G]) representing significantly less HI than the homozygous c.235delC. The group with c.[235delC]+[176_191del16] represented more severe HI than the reference group.
In the groups with only one subject, Fisher's exact probability testing was applied to compare the median PTA0.5-4 kHz with the reference group (Table 1).Though P values did not imply significant difference with the homozygous c.235delC group, the conclusion could not be drawn certainly because of the small sample size.
Discussion
Correlation between GJB2 genotypes and phenotypes
Since the GJB2 gene plays an important role in the etiology of NSSHI, it was preferred to the primary screening gene for newborns. Several multicenter studies looking at the correlation between GJB2 mutations and the hearing levels have been performed, and some valuable conclusions were drawn. That is, the homozygous c.[30_35delG] genotype was frequently associated with profound HI, and the genotypes of both inactivating mutations were significantly associated with profound and severe HI. Further, the subjects with genotypes comprising at least one noninactivating mutation or two showed obviously moderate or mild hearing loss (Cryns et al., 2004; Snoeckx et al., 2005).
Some results indicated that the level of HI usually depends on the less severe mutation, which meant that patients presenting with significantly milder hearing loss carried at least one non-inactivating mutation. This point was really supported by some studies, for example, c.[30_35delG]+[109G>A], c.[368C>A]+[368C>A], c.[235delC]+[109G>A], c.[30_35delG]+[-23+1G>A], and c.[30_35delG]+[101T>C] were demonstrated to associate with milder clinical symptoms. In the study sample, some subjects with such genotypes had the PTA level of 35-70 dB (Cryns et al., 2004; Marlin et al., 2005; Snoeckx et al., 2005).
Though the GJB2 gene is the most frequent causative gene in China, the relationship between the phenotypes and the genotypes has not been reported (Wang et al., 2004). Our study first did this research in a large cohort in Chinese population. As shown in the results, we found three genotypes present in more than two observations to be associated with significantly less severe HI than the homozygous c.235delC group; so, our data also supported the popular opinion. Both the mild genotypes of ([p.Val37Ile]+[p.Val27Ile; p.Glu114Gly] and homozygous [p.Val27Ile; p.Glu114Gly]) were in the NT+NT category.
In addition, we found that one genotype (c.[235delC]+[176_191del16]) was associated with more significantly severe HI than the homozygous c.235delC group. In Caucasians, c.[30_35delG]+[del(GJB6-D13S1830)]was found with more severe HI than the homozygous c.35delG group (Snoeckx et al., 2005).
There was no significantly different HI between some genotypes and the homozygous c.235delC group, which were c.[30_35delG]+[299_300delAT], c.[235delC]+[30_35delG], c.[299_300delAT]+[176_191del16], c.[235delC]+[30_35insG], c.[235delC]+[504_505insAAGG], and c.[299_300delAT]+[504_505insAAGG]. It was noted that the P value of homozygous c.[299_300delAT] was 0.064, which showed a tendency to distinguish from the reference group. The result was similar to that in the Caucasian population to some extent, though the map of GJB2 mutations was different between the two different populations. If we can deduce that the HI level is related to some specific genotypes, it would be useful to examine the effect of environmental factors, such as infections or medications, and genetic background on hearing loss increase in patients with DFNB1 (Wilcox et al., 2000).
A phenomenon emerged that a large scatter of the degree of hearing loss existed in the three genotypes which had the largest number of patients tested, as seen in the other study (Cryns et al., 2004). The simplest explanation for the difficulty in correlating a specific genotype to clinical symptoms is that additional genetic-, epigenetic-, and/or environmental factors could modify the phenotype. It was further supported that some cases of severe or profound NSSHI are associated with a single GJB2 mutation (data not shown).
Though some studies showed that no mutations in GJB2 were found to relate to postlingual deafness in Japanese, Jews, and the French (Denoyelle et al., 1999; Kudo et al., 2000; Sobe et al., 2000), other studies found that p.Leu90Pro, p.Met34Trp, and p.Trp44Cis may have correlation with postlingual progressive deafness in Poland, Australia, and America, respectively (Kenneson et al., 2002; Pollak et al., 2007). It was unclear whether the progression existed in truncating genotypes. In our study, we found 11 subjects to be with postlingual deafness. As shown in results, the genotypes scattered in NT+NT and T+T groups, even in four subjects with homozygous c.[235delC] genotype. Our study provided new proof of postlingual deafness caused by the GJB2 truncating mutation. It seems contradictory with the putative molecular pathogenic effect of truncating mutation, which could somewhat be explained as the pathogenic fluctuation or progress, though no relationship was discovered between age and HI so far (Cryns et al., 2004). This puzzle requires thorough cellular or animal model study of GJB2 mutations (Choi et al., 2009).
Contradictory outcomes between functional research and phenotype
GJB2 codes the Cx26 protein family, which connects the gap junction to form homotypic or heterotypic channels to maintain tissue homeostasis and to allow fast intercellular electrical communication. An extensive network of gap junctions is found in two kinds of cells: the cochlear nonsensory epithelial cells and the cochlear connective tissue cells. Cx26 can form homomeric and homotypic channels. Cx26 and Cx30 have been demonstrated to form heteromeric gap junctions in coexpression studies in Hela cells, and it has also been demonstrated that certain Cx26 mutations exert a dominant negative effect on Cx30, by affecting Cx30 channel permeability (Marziano et al., 2003).
Plenty of functional studies have been performed to study the Cx26 protein and the channels the CX26 participated in. Pathogenic changes of different mutations in GJB2 varied, which interfered with the translation or stability of Cx26 protein (p.Met1Val, p.Arg184Pro), or disturbed protein trafficking and assembling in hemi-channels (p.Leu90Pro, c.[30_35delG], c.[235delC]), or disabled formation of homotypic gap junction channels (p.Val37Ile, p.Trp77Arg, p.120delGlu, p.Met163Val, and p.Met34Thr). Some mutations were not thoroughly studied in vitro (c.[559_604dup46], c.[176_191del16], p.Arg32Cis) or were thought to have no effect on protein function (c.[341A>G], p.Ile203Thr) (Hoang Dinh et al., 2009).
However, some functional outcomes were discordant with the phenotype. Though the mutation of c.[-23+1G>A] did not yield any detectable Cx26 protein and was similar to the mutation of c.[30_35delG], the subjects with c.[30_35delG]+[-23+1G>A] genotype had significantly less severe HI comparing with c.35delG homozygotes (Cryns et al., 2004).
In this study, we found the mutation p.Val37Ile to be associated with less severe HI compared with c.235delC homozygotes as well in other studies (Lin et al., 2001; Snoeckx et al., 2005). However, some expression functional studies have demonstrated complete loss of channel activity in cells transfected with p.Val37Ile (Kudo et al., 2000; Bruzzone et al., 2003), whereas some trials confirmed it was not pathogenic (Hwa et al., 2003; Wattanasirichaigoon et al., 2004). The same contradiction between functional study and HI level also emerged in the study of p.Leu90Pro and p.Met34Tro.
An explanation for this phenomenon was that the p.Val37Ile and p.Met34Thr had lower penetrance compared with mutations of undisputed pathogenicity (Pollak et al., 2007). Another explanation was that these studies mainly focused on the channel properties. The effect of p.Val84Leu on HI was discovered to be another novel way of impairing the permeability of IP3, which was essential in mediating the calcium ion propagation in cochlear supporting cells (Thonnissen et al., 2002), and suggested that the complicated effect of GJB2 mutations in HI should be studied in a model similar to in vivo conditions.
Combined causative mutation of p.Val27Ile(c.79A>G) and p.Glu114Gly(c.341G>A)
In the Chinese population, p.Val27Ile and p.Glu114Gly mutation were frequent, and both were thought to be the polymorphism by majority. However, p.Val27Ile has low frequency (less than 2%) in Caucasians (Roux et al., 2004). p.Val27Ile locates in a transmembrane part, and p.Glu114Gly situates in the intracellular loop. The cellular functional studies showed that p.Glu114Gly had no effect on protein, but p.Val27Ile caused dysfunction of protein in vitro (Choung et al., 2002; Bruzzone et al., 2003). A recent study reported in the 30th ARO Annual MidWinter Research meeting discovered that dye transfer and calcium conductance was nil in the homozygous [p.Val27Ile; p.Glu114Gly] injected cells; whereas a compound heterozygote, p.Glu114Gly homozygote, and a p.Glu114Gly heterozygote showed impaired intercellular biochemical coupling (Pilipenko et al., 2007).
The combination of p.Val27Ile and p.Glu114Gly was identified as one causative mutation (Putcha et al., 2007). There were a total of 106 alleles (18.5%) of [p.Val27Ile; p.Glu114Gly] among deaf subjects in our study. We screened this mutation in 400 newborns and found that only 5 subjects present with it (0.6%). [p.Val27Ile; p.Glu114Gly] appeared far more frequently in the deaf group than in the normal group. The frequency of this controversial mutation in the deaf group was 11.7% in the Altai population and 11.3% in Singapore (Posukh et al., 2005). Our epidemiological data sustained that [p.Val27Ile; p.Glu114Gly] had very close correlation with deafness.
Mechanism of impairment
The GJB2 mutations primarily affected outer hair cells, because the impairment (detectable by otoacoustic emissions) seems to be constant in all genotypic groups and even in the subjects carrying only one GJB2 mutated allele. Our DPOAE data supported this opinion, for we found that normal waves could not be elicited in patients whose thresholds were lower than 60 dB. The impairment was supposed to be caused by local intoxication of the organ of Corti by the accumulating K+ ions, which would affect the function or survival of hair cells (Lefebvre and Van De Water, 2000). An alternative opinion was that interruption of K+ ions circulation would prevent the development of the receptor potential in hair cells (Kikuchi et al., 2000). Inner hair cells and nerve impairment were found to be variable in the distinct genotypic combinations analyzed (Engel-Yeger et al., 2003). Another two theories exist that explain the causative mechanisms of CX26 mutations, including a theory of endothelial barrier breakage and a theory of deficiency in gap-junction protein-facilitated metabolite transportation (Hoang Dinh et al., 2009).
Footnotes
Acknowledgments
We thank the patients and their families for their cooperation during this work. This work was supported by grants from the National Natural Science Foundation of China, Key Project (grant no. 30830104), the National Natural Science Foundation of China (grant nos. 30672310, 30771203, and 31071166), Beijing Nature Science Technology Major Project (grant no. 7070002), Natural Science Foundation of Guangdong Province, China (grant no. 8251008901000007), and Science and Technology Planning Project of Guangdong Province (grant no. 2009A030301004).
Disclosure Statement
No financial conflict of interest relevant to this article was reported.