
Abstract
Mutations at splicing consensus sequences have been shown to induce splicing errors such as exon skipping or cryptic splice site activation. Here, we identified eight splicing products caused by a G-to-T transversion mutation at the splice acceptor site of exon 14 of the dystrophin gene (c.1603-1G>T). Unexpectedly, the most abundant product showed skipping of the two consecutive exons 14 and 15, and exon 14 skipping was observed as the second most abundant product. To examine the cause of this splicing multiplicity, minigenes containing dystrophin exons 14 and 15 with their flanking introns were constructed and subjected to in vitro splicing. Minigenes with the wild-type sequence or a G>A transition at position c.1603-1 produced only the mature mRNA. On the other hand, the minigenes with a G>T or G>C transversion mutation produced multiple splicing products. A time-course analysis of the in vitro splicing revealed that splicing of the middle intron, intron 14, was the first step in transcript maturation for all four minigene constructs. The identity of the mutant nucleotide, but not its position, is a factor leading to multiple splicing outcomes. Our results suggest that exon skipping therapy for Duchenne's muscular dystrophy should be carefully monitored for their splicing outcomes.
Introduction
Duchenne's and Becker muscular dystrophy (DMD/BMD) are the most common forms of inherited myopathy and are caused by mutations in the dystrophin gene. DMD is a rapidly progressive disease that usually results in the death of patients in their twenties, whereas BMD is a clinically less severe form of the disease and often has only a slightly debilitating effect. The difference between DMD and BMD can be explained by the reading frame rule: frame-shift mutations that generate premature stop codons in the coding sequence of dystrophin mRNA usually result in the DMD phenotype, whereas mutations that maintain the original reading frame cause the milder BMD phenotype (Monaco et al., 1988). Therefore, it is important to consider how splice site mutations could alter the splicing products of the dystrophin gene, and thus whether they will lead to either the DMD or the BMD phenotype. For this reason, dystrophinopathy is one of the most extensively studied diseases related to splice site mutations and their resultant products (Matsuo et al., 1991; Nishiyama et al., 2008).
The dystrophin gene is the largest human gene, spanning 2500 kb on the X chromosome. It has a complex structure, including a large number of exons, long introns, and several alternative promoters (Den Dunnen et al., 1989; Ahn and Kunkel, 1993; Nishio et al., 1994). Dystrophin pre-mRNA splicing would seem to require extraordinarily strict regulation. However, single-nucleotide mutations in splice acceptor or donor sites have been reported to cause splicing alterations, including exon skipping, cryptic splice site activation, or both (Hagiwara et al., 1994; Tuffery-Giraud et al., 1999; Adachi et al., 2003; Thi Tran et al., 2005; Habara et al., 2009). Despite the identification of a large number of splice site mutations in many diseases, there is still no established way to predict what splicing patterns these mutations will produce (Wimmer et al., 2007; Hertel, 2008).
In this study, we identified multiple splicing outcomes caused by a transversion mutation at the splice acceptor site of dystrophin exon 14. In in vitro splicing assays, transversion but not transition mutations induced multiple splicing outcomes. We suggest that the manipulation of splicing with antisense oligonucleotides (AOs) should be carefully evaluated in DMD treatment.
Materials and Methods
Genomic DNA analysis
Genomic DNA was extracted from a BMD male (KUCG557) and a control as previously described (Matsuo et al., 1990). A mutation in the dystrophin gene was sought by direct sequencing (Nishiyama et al., 2008; Takeshima et al., 2010).
Dystrophin mRNA analysis
Dystrophin mRNA in skeletal muscle was analyzed as previously described (Roberts et al., 1991; Surono et al., 1999). A fragment spanning exons 13 to 18 was amplified by reverse transcription-polymerase chain reaction (RT-PCR) using a set of primers (c13f; 5′-GCTGCTTTGGAAGAACAACTT-3′ and 1F; 5′-CTTCTGAGCGAGTAATCCAGCT-3′) using conditions as previously described (Habara et al., 2009). The amplified products were sequenced after subcloning, as previously described (Thi Tran et al., 2005).
In vitro splicing analysis
In vitro splicing analysis was conducted using the preconstructed minigene of the expression vector H492 that encodes two cassette exons (exons A and B) and a multicloning site in the intervening sequence (Habara et al., 2008).
A region spanning intron 13, exon 14, intron 14, exon 15, and intron 15 was PCR amplified from genomic DNA of the index case and a control using the primer set: 5′-GCGGCTAGCTCAGAAAGAGTGTCCCTTCCAA-3′ and 5′-GCGGGATCCCACTTTAATTCAGAAAAGTAGCAA-3′. Amplified products were digested with NheI and BamHI restriction enzymes (New England Biolabs, Ipswich, MA) and inserted into the H492 vector that was predigested with the same enzymes. Two mutant constructs, c.1603-1G>A and c.1603-1G>C, were prepared by means of overlap extension PCR and were inserted into the H492 vector. After checking their sequences, the resultant plasmids were transfected into HeLa cells for splicing assays as previously described (Habara et al., 2009). The cells were harvested after 24 h of incubation for extraction of total RNA.
Time-course analysis
To determine the splicing order of the introns, in vitro splicing was stopped after 1, 2, 3, or 4 h by extracting total RNA. The resulting splicing products were analyzed as previously described. RT-PCR products were semiquantified using a DNA 1000 LabChip kit on an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) and their sequences were determined by subcloning and sequencing.
Results
A G-to-T transversion mutation was identified at the 3′ end of intron 13 of the dystrophin gene (c.1603-1G>T) in a Japanese BMD patient (Fig. 1). Although the mutation was located within an intron, the mutation changed the highly conserved AG dinucleotides at the splice acceptor site to AT, thereby decreasing the splice site strength (Ri) of the splice acceptor site from 12.3 to 3.5. It was strongly expected that skipping of the downstream exon 14 would be the splicing outcome of this mutation. However, dystrophin mRNA expressed in the patient's skeletal muscle was found to contain multiple splicing products. When a region spanning exons 13 to 18 was amplified by RT-PCR, four amplified bands were visualized on agarose gels, with different densities from clearly visible to barely visible (Fig. 1). Unexpectedly, subcloning and sequencing revealed that the most abundant product lacked not only exon 14 but also exon 15, indicating the skipping of two consecutive exons (Fig. 1). The second most abundant product corresponded to the expected product that lacked only exon 14. As these two abundant mRNAs maintained the original dystrophin reading frame, the index case was diagnosed as BMD at the mRNA level. This matched with his clinical phenotype.
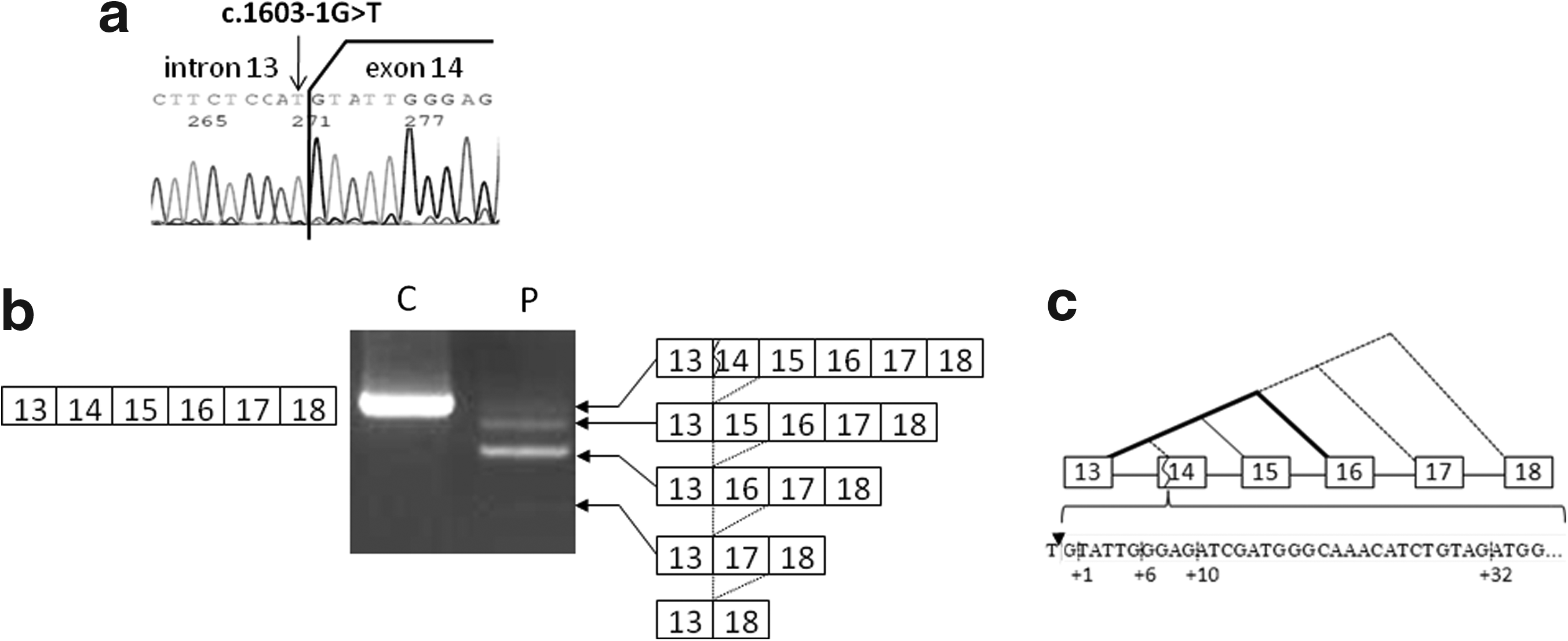
Genomic and mRNA analysis of the dystrophin gene.
The third faintly visible product, which corresponded to the wild-type dystrophin size, maintained all six exons from exons 13 to 18. However, subcloning and sequencing of this band revealed four different clones, containing a deletion of 1, 6, 10, or 32 nucleotides at the 5′ end of exon 14 (Fig. 1). It was unclear whether these were genuine splicing products or artifacts. Examination of the genomic sequences around these unexpected deletions revealed that the ends of two of the deletions, at the 10th and 32nd nucleotides, comprise AG dinucleotides, whereas the ends of the other two, at the 1st and 6th nucleotides, comprise TG dinucleotides (Fig. 1). Because all these four sites scored as potential splice acceptor sites, it was concluded that all four clones were derived from alternative splicing products, activating a cryptic splice acceptor site located +1, +6, +10, or +32 nucleotides from the intron 13/exon 14 boundary (Fig. 1). The smallest, barely visible band lacked exons 14, 15, and 16 (Fig. 1) and clones lacking exons 14, 15, 16, and 17 were also identified. Eight splicing patterns had been identified in the patient's muscle RNA: four patterns of exon skipping and four patterns of cryptic splice site activation (Fig. 1).
Multiple splicing outcomes caused by a single-nucleotide change at the splice acceptor site of the mammoth dystrophin gene were unexpected. This suggested that the mutation identified in the index case exerts a strong influence on the splicing machinery. To demonstrate this, an in vitro splicing system using the H492 minigene was employed. Fragments encompassing exons 14-15 and their flanking introns, with wild-type or mutant (c.1603-1G>T) sequence, were amplified from genomic DNA by PCR and inserted into the multicloning site within the intron between exons A and B of the H492 minigene (Fig. 2a). Constructed minigenes were transfected into HeLa cells and the resulting splicing products were analyzed by RT-PCR after 24 h. From the wild-type construct, a single amplified product was obtained and was confirmed to be the expected mature mRNA consisting of exons A, 14, 15, and B (Fig. 2). This in vitro system was therefore confirmed to effect accurate splicing of dystrophin exons 14 and 15.
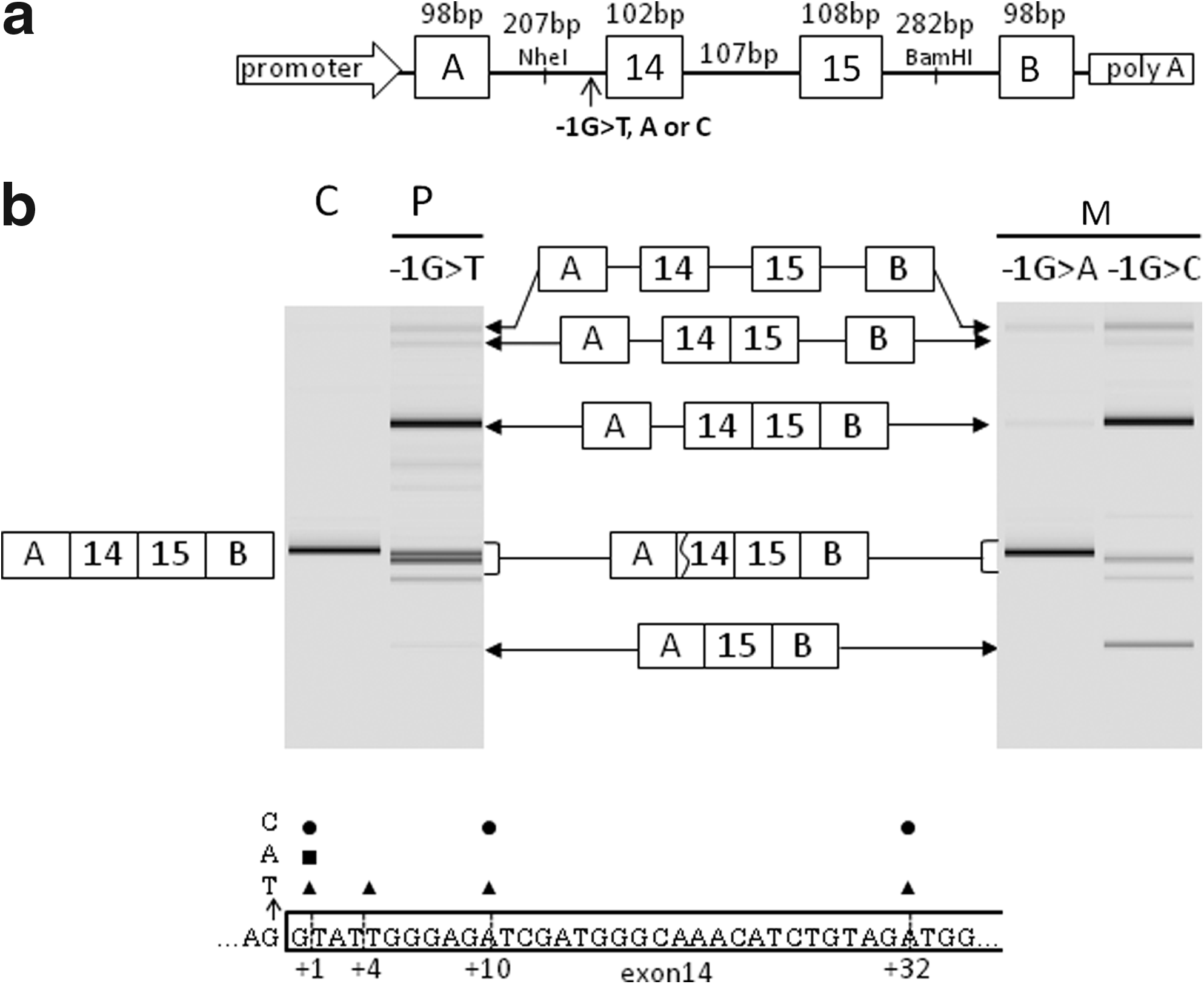
In vitro splicing analysis.
For the mutant minigene harboring the c.1603-1G>T transversion, multiple RT-PCR amplification products were identified: one major band, two minor bands, and one weak band (Fig. 2). Sequencing of the major band revealed an intermediate splicing product that lacked both introns 14 and 15 but retained intron A unspliced. This indicated that the splicing machinery recognized the two downstream introns, but not the upstream intron A in the presence of the c.1603-1G>T change. This outcome was compatible with the disruption of the splice acceptor site of intron A by the mutation. By subcloning and sequencing of the two minor products and the one weak product, four clones were identified, which differed in their exon 14 5′ end sequences in the mature mRNA. Each had a 1-, 4-, 10-, or 32-nucleotide deletion from the 5′ end of exon 14 (Fig. 2); the 10-bp deletion was the most common. Although activation of four cryptic splice acceptor sites within exon 14 was shown in vitro as well as in vivo, only three were common to both. In vivo, the sixth nucleotide was activated as a cryptic splice site (TG), whereas in vitro, the 4th nucleotide was activated (AT). Remarkably, pre-mRNAs maintaining all three introns and mRNA lacking only intron 14 were identified. Because the pre-mRNA was not detected for the wild-type minigene, the G>T mutation was considered to affect the splicing reaction fundamentally. However, skipping of exons 14 and 15, or the single exon 14, was not observed in vitro. It was unclear why the expected exon skipping did not occur, even though multiple splicing outcomes caused by the G>T transversion mutation were exemplified in vitro (Fig. 2).
We questioned whether nucleotide changes other than the G>T transversion located at an identical position would also produce multiple splicing outcomes. Minigenes with substitution of G to A or C were constructed and subjected to the same in vitro splicing assays. Unexpectedly, the minigene with the G>A transition mutation produced one clear band (Fig. 2). Interestingly, this product corresponded to the mature mRNA but had a deletion of a G nucleotide at the 5′ end of exon 14. This G nucleotide was probably recognized as the last nucleotide of intron A, resulting in a simple splicing outcome. This minigene also produced a faint band corresponding to pre-mRNA; this was probably due to disturbance of the entire splicing reaction (Fig. 2).
Remarkably, the minigene with the G>C transversion mutation produced many products: one major band, four additional weak bands, and one barely discernible band. The most abundant band corresponded to a splicing intermediate that removed two downstream introns (introns 14 and 15) but retained intron A (Fig. 2). The second abundant, smallest product had the exon structure A-15-B, indicating exon 14 skipping. In addition, three transcripts were identified with 1-, 10-, or 32-nucleotide deletions at the 5′ end of exon 14 of the mature mRNA (Fig. 2), indicating three cryptic splice site activations. The G>C transversion mutation induced multiple splicing outcomes. These results indicated that a single-nucleotide change at the splice acceptor site changed the splicing pathways, but the splicing outcomes per se were dependent on the particular substituted nucleotide.
To clarify the splicing process in transversion or transition mutations at c.1603-1, the time course of the splicing reactions in the minigenes was investigated 1-4 h after transfection. After 1 h, an intermediate product that lacked intron 14 was detected for all four minigenes, in addition to the pre-mRNA (Fig. 3). This indicated that the first step of splicing occurs at the middle intron (intron 14), even in the presence of a normal splice acceptor site for intron A. After 2 h, the mature mRNA constituted >60% of the total mRNA for the wild-type minigene and 55% for the G>A mutant minigene, indicating an uneventful splicing reaction. In contrast, mature mRNA constituted only 10%-20% for minigenes with the G>T or G>C transversions. This indicated that splicing of intron A in the context of a G>T or G>C transversion was strongly inhibited. After 3 h, >80% of the total transcripts were mature mRNA for the wild-type minigene or for the minigene with the G>A transition. However, mature mRNA with activated cryptic splice sites within intron A occupied <40% for the minigenes with the G>T or G>C transversion. This indicated that the last step, the splicing of intron A, was strongly hampered in the presence of a transversion mutation.

Time-course analysis of minigene splicing reactions.
After 4 h, the splicing reaction was almost complete, with mature mRNA constituting >90% of the total transcripts for the wild-type minigene or the minigene with the G>A mutation. For minigenes with the G>T or G>C transversion, mature mRNA comprised less than half of the total. We considered this to be due to severe disturbance of the splicing of intron A in the presence of a transversion mutation.
In summary, we found that transversion mutations led to multiple splicing outcomes by suppressing splicing reactions. Our data suggest that the identity of the mutant nucleotide can dramatically influence splicing outcome.
Discussion
In this study, a transversion mutation from G to T (c.1603-1G>T) at the splice acceptor site of exon 14 of the dystrophin gene was shown to induce multiple splicing outcomes in a Japanese BMD case. Altogether, eight splicing patterns for dystrophin mRNA were identified in the patient's muscle (Fig. 1). We questioned why this wide variety of splicing patterns was produced from a single-nucleotide change.
In the dystrophin gene, 80 donor and 137 acceptor splice site mutations have been reported (www.dmd.nl/nmdb/home.php). Most of these showed skipping of an adjacent exon as the outcome of the mutation, and some resulted in cryptic splice site activation (Bartolo et al., 1996; Adachi et al., 2003; Thi Tran et al., 2005). Skipping of multiple exons has been reported only rarely: among the reported splice site mutations of the dystrophin gene, only six have been described to induce skipping of multiple exons (Tuffery-Giraud et al., 2004, 2005; Deburgrave et al., 2007; Taylor et al., 2007; Takeshima et al., 2010). Of these six, three were splice acceptor site mutations including the present case. Three were splice donor site mutations, but two of these were an identical mutation at the splice donor site of exon 29 (Deburgrave et al., 2007). It was supposed that splice acceptor site mutations have a greater tendency to promote multiple exon skipping. It is remarkable that mutations at the splice donor site of exon 29 and at the splice acceptor site of exon 13 have each been reported twice to cause multiple exon skipping. This suggests that these two exons predispose to multiple exon skipping in the presence of a splice site mutation.
In the index mutation, we supposed that the double exon skipping was due to the short length of intron 14 (107 bp). However, in vitro splicing of the minigene with an intact intron 14 failed to produce double exon skipping (Fig. 2). This indicated that the short length of intron 14 is not the sole determinant of multiple exon skipping. Remarkably, the time-course analysis of minigene splicing revealed that splicing of the middle intron, intron 14, occurred first for all four minigenes (Fig. 3). Because the structure of the intron and neighboring exons was accurate in the minigenes, we presume that intron 14 is spliced out at an early stage also in vivo. It is supposed that, in the patient's muscle, the removal of intron 14 is the major splicing event in the early stages, and then the splicing machinery identifies the splice acceptor site of exon 16 and the donor site of intron 13, thereby promoting the skipping of the two consecutive exons 14 and 15. As a minor pathway, the splicing machinery identifies the splice acceptor site of exon 15 and the donor site of intron 13, leading to exon 14 skipping.
Multiple splicing outcomes were exemplified for minigenes with transversion mutations but not for the minigene with a transition mutation (Fig. 2). Although a multiplicity of splicing outcomes was demonstrated in vitro as in vivo, the actual splicing patterns were different: a splicing intermediate retaining intron A was a major product in vitro (Fig. 2), whereas exon-skipped mature mRNAs were the main products in vivo (Fig. 1). Among the four in vitro splicing reactions, only the minigene with the G>C mutation produced exon 14-skipped mRNA. Even though the two transversion mutations both exerted drastic changes (Fig. 2), each produced different splicing outcomes. It is difficult to explain this difference. We conclude that both transversion mutations cause drastic changes in the splicing machinery and multiple splicing outcomes, but the identity of the substituted nucleotide induces the splicing pattern differences.
Recently, the manipulation of dystrophin pre-mRNA splicing is attracting much attention as a way to treat DMD (Matsuo, 1996; van Deutekom and van Ommen, 2003). AOs against splicing regulatory elements have been shown to induce exon skipping, thereby enhancing dystrophin expression in DMD patients (Takeshima et al., 2006; van Deutekom et al., 2007). Our results flag the possibility of multiple splicing outcomes after manipulation of splicing with AOs. Particular caution should be exercised when inducing skipping of the exons that have been reported to induce multiple exon skipping in the presence of a splice site mutation (Tuffery-Giraud et al., 2004, 2005; Deburgrave et al., 2007; Taylor et al., 2007; Takeshima et al., 2010). Considering that even a single-nucleotide change within exon sequence has been shown to induce splicing error (Shiga et al., 1997; Nishiyama et al., 2008), AO is supposed to induce unwanted splicing outcomes, such as activation of a cryptic splice site created by a new junction. In establishing exon skipping therapy with AO, therefore, splicing outcomes should be carefully monitored.
Footnotes
Acknowledgments
The authors thank Professor Peter K. Rogan for his encouraging support and Ms. Kanako Yokoyama for her secretarial help. This work was supported in part by a Grant-in-Aid for Scientific Research (B) and Grant-in-Aid for Exploratory Research from the Japan Society for the Promotion of Science; a Health and Labor Sciences Research Grant for Research on Psychiatric and Neurological Diseases and Mental Health; and a research grant for Nervous and Mental Disorders from the Ministry of Health, Labor, and Welfare, Japan.
Disclosure Statement
No competing financial interests exist.