
Abstract
Duchenne's muscular dystrophy and Becker muscular dystrophy (BMD) are X-linked recessive disorders caused by mutations in the dystrophin gene. In this project, 100 unrelated male patients were initially screened for deletions in the dystrophin gene by multiplex polymerase chain reaction, of whom 52 were positive. We performed the multiplex ligation-dependent probe amplification (MLPA) method on 43 of the remaining 48 patients, as well as 12 females suspected to be carriers, to detect deletions and duplications of their dystrophin gene. The MLPA method found deletions and duplications in 8 unidentified male patients. Sequencing revealed that in one case the deletion detected was a point mutation. One of 12 females was heterozygous for deletion of exons 49 and 50. In conclusion, the MLPA method proved to be reliable for studying affected males as well as female carriers.
Introduction
D
DMD is the most common muscle disease in children, with an incidence of around 1 in 3500 live born males, and the incidence of BMD is around 1 in 18,500 live born males (Lai et al., 2006).
DMD is a more severe disease with delayed walking in early childhood, pseudohypertrophy of muscles, an increased creatine kinase level, and progressive deterioration in muscle power, and most patients become confined to a wheelchair by the age of 12 and die early in their third decade of life from respiratory or cardiac failure (Lai et al., 2006).
BMD is a milder phenotype with age of onset around 12 years and a slower clinical progression (Fai-Man Lo et al., 2006). Some patients have no symptoms until much later in life. Loss of ambulation also varies from adolescence onward, with death usually in the fourth or fifth decade (Hoffman et al., 1987; Den Dunnen et al., 1992; Nobile and Marchi, 1994).
The dystrophin gene, located at Xp21, is the largest human gene known. It consists of 79 exons and has a 2.4 Mb length. Its mRNA transcript is 14 kb long and is expressed in skeletal muscle and brain. The dystrophin protein is a 427-kDa sarcolemmal protein that is involved in the linkage between the extracellular matrix and the cytoplasmic cytoskeleton (Yoshida and Ozawa, 1990; Ervasti and Campbell, 1991, 1993).
In DMD patients, no dystrophin protein is produced, whereas in BMD patients the amount of dystrophin protein decreases to 10%-40% or a partly functional dystrophin protein with an altered size is produced (Lai et al., 2006; van Essen et al., 1997).
Partial gene deletions (∼60%), duplications (∼5%), or point mutations and deletions/insertions of few nucleotides (35%) account for the DMD/BMD patients (Traverso et al., 2006; Forrest et al., 1987; Hu et al., 1988, 1990). Mutations may be inherited from asymptomatic female carriers (70%) or occur de novo (30%). Therefore, detecting carriers and prenatal diagnosis prevent the birth of new affected cases (Traverso et al., 2006; Worton and Thompson, 1988).
Moreover, appropriate treatment can be applied according to the specific dystrophin gene mutation. For example, PTC124 selectively induces ribosomal readthrough of premature but not normal termination codons (Welch et al., 2007). Antisense therapy uses small oligonucleotides to correct frame-shifting or nonsense mutations by skipping a mutated exon or exons that disrupt the reading frame. This results in the expression of a truncated but at least partially functional dystrophin (Lu et al., 2005). Also, oligonucleotide-mediated gene editing corrects single point mutation or restores the reading frame of the dystrophin gene (Bertoni and Rando, 2005).
It is possible to confirm the presence of dystrophinopathy and differentiate between DMD and BMD by immunohistochemical staining of muscle biopsies. However, not all the patients will give muscle biopsies and also dystrophin staining is not always conclusive in BMD (Lai et al., 2006; van Essen et al., 1997).
There are different methods for detection of deletions/duplications in the dystrophin gene. Three sets of multiplex polymerase chain reaction (PCR) developed by Beggs, Chamberlin, and Kunkel allow a detection of 90%-95% of the deletions in male patients (Beggs et al., 1990; Chamberlain et al., 1988; Kunkel et al., 1991). However, just some exons are covered in this method and the exact breakpoint site of most deletions remains unknown. On the other hand, a multiplex PCR method does not detect duplications in males as well as deletions/duplications in carrier females. Quantitative southern blotting is a reliable method for detecting duplications and identifying carrier females. However, it is not always able to detect small deletions; moreover, this method is time consuming, requires several hybridization steps, and is not ideal as a routine technique (Schwartz and Dunø, 2004; Den Dunnen et al., 1989; Yamagishi et al., 1996). Using the fluorescent-labeled primers in quantitative PCR, detection of duplications and identification of carriers of deletions and duplications are possible but not all the exons are covered in this method; so deletions and duplications outside the hotspots will be missed (Yau et al., 1996). The multiplex, amplifiable probe hybridization method allows the detection of deletions and duplications of all 79 exons of the dystrophin gene and is suitable for carrier diagnosis, but it is difficult and laborious to use (White et al., 2002; Janssen et al., 2005).
Fluorescence in situ hybridization (FISH) is another method to perform, although it is not suitable for studying small fragments. Moreover, it cannot be used on purified DNA (Schouten et al., 2002; Klinger et al., 1992).
The multiplex ligation-dependent probe amplification (MLPA) method allows the relative quantification of up to 40 different nucleic acid sequences in a single reaction tube (Schouten et al., 2002; Gatta et al., 2005). The method is accurate and reliable for identifying deletions and duplications in both males and females (Schwartz and Dunø, 2004).
In brief, each MLPA probe consists of two oligonucleotides that hybridize to the immediately adjacent target sequences. The probes have a nonhybridizing stuffer sequence with different length (19-364 nt), making it possible to differentiate between different PCR products. After an overnight hybridization to the target DNA, the right and left probe oligonucleotides are joined by a ligation reaction. As the ligation products have both forward and reverse PCR primer binding sites, it is possible to amplify them by PCR. The different lengths of the products are separated on a capillary sequencer/fragment sizer, and the peak areas or the peak heights are quantified and the copy numbers of the exons are estimated (Schouten et al., 2002; Kozlowski et al., 2008).
In this study, we used the MLPA method for detecting deletions and duplications in DMD/BMD patients whose deletions were not detected by multiplex PCR designed for 27 exons and the muscle promoter (Dp427m) of the dystrophin gene. Also, MLPA was used for carrier detection. To our knowledge, this is the first report from Iran.
Materials and Methods
Patients
In total, 100 unrelated male patients (76 DMD and 24 BMD patients) were referred to our laboratory by the neurology specialists. Diagnosis was based on clinical signs, electromyogram, and high serum creatine kinase activity. Consent was obtained from all patients for carrying out research on their specimens.
Multiplex PCR was performed on all the patients and was capable to find deletions in 52 patients.
As we did not have access to DNA samples from 5 of 48 unidentified patients, MLPA was not performed for them.
Forty-three patients who had tested negative by multiplex PCR and also 8 male patients with known deletions as positive control samples were studied by the MLPA method.
In addition, we studied 12 unrelated females for carrier detection. The short tandom repeats (STRs) were either not informative for these females following linkage study performed to identify their chromosome phase or the probands were not alive in their families.
DNA extraction
DNA was extracted from peripheral blood samples by the salting-out method. For the MLPA candidate samples, an ethanol precipitation step was added to further purify DNA samples using the protocol written at MRC-Holland web site (www.mrc-holland.com/).
Multiplex PCR
Deletions in the dystrophin gene was examined utilizing four sets of multiplex PCR covering the exons in the deletion hot spots on 100 unrelated male patients adopting protocols recommended at the Leiden Muscular Dystrophy pages (www.dmd.nl/) (Table 1).
Multiplex ligation-dependent probe amplification
The MLPA reactions (two reactions for each sample) were performed on DNA samples. In each MLPA run, four DNA samples of normal people, one DNA sample of a patient with a known deletion, and one sample with no DNA (blank) were included as control samples for normalization, positive control, and negative control, respectively.
The MLPA DMD test kit (SALSA P034-A2/P035-A2) was obtained from MRC-Holland (Amsterdam, The Netherlands). This kit contains 79 probes for the DMD exons and 1 probe for the alternative exon 1: DP427C.
Briefly, 100-150 ng of genomic DNA in 5 μL TE was heated to 98°C for 20 min. Then, 3 μL hybridization master mix containing MLPA buffer and probe mix was added following the reduction of temperature to 25°C. The reaction mixture was denatured at 95°C for 2 min and incubated for 16-17 h at 60°C. After hybridization reaction, a ligase master mix (32 μL) was added and incubated at 54°C for 15 min for the ligation step. The ligase enzyme was inactivated by heating to 98°C for 5 min. Then, a fluorescent multiplex PCR was performed (35 cycles: 30 s at 95°C, 30 s at 60°C, and 60 s at 72°C, followed by incubation at 72°C for 20 min).
Fragment separation
About 0.7 μL PCR product was mixed with 0.2 μL GS-500 LIZ size standard and 9 μL HiDi formamide and this mixture was heated to 94°C for 5 min. The fragments were separated on an ABI model 3130 capillary sequencer using POP4 or POP7 as polymer. The initial settings for fragment separation are shown in Table 2.
MLPA analysis
For the analysis of raw data, we used Gene marker software. The sizes of exon-specific peaks were identified according to their migration relative to the GS-500 LIZ size standard. Population normalization was chosen as MLPA normalization method and peak ratios were calculated by the area of the peaks. The average ratio of the peak area of the exons after normalization is shown in Tables 3 and 4.
In male patients, the ratio for deleted exons was considered 0, although any normalized ratio below 0.4 was considered a possible deletion and the related exon was tested by PCR and sequencing.
Confirmation of deletions and duplications of single exons
Identified deletions and duplications were considered reliable when they involved two or more adjacent exons and no further investigations were performed. However, as we did not have any control sample for duplication, we confirmed duplication in one patient by real-time PCR.
The absence of only one peak related to a single exon in males was investigated further using PCR primers flanking that exon.
If a deletion could not be confirmed by PCR, the PCR products of the related exons were sequenced to detect the probable point mutations within the exons, which might destroy or considerably reduce the annealing of the MLPA probes.
Results
Fifty-two of 100 patients showed deletions by multiplex PCR method. As DNA samples were not available for 5 of 48 unidentified cases, MLPA was carried out on 43 patients.
MLPA was able to detect all known deletions previously identified by multiplex PCR. Also, as the MLPA probes are designed for all exons of dystrophin gene, in some cases it revealed larger deletions when compared with the multiplex PCR findings. Table 5 compares the multiplex PCR and MLPA results for eight positive control samples.
Sample 6 was not initially tested for exons 54 and 55 in the multiplex PCR.
MLPA, multiplex ligation-dependent probe amplification; PCR, polymerase chain reaction.
Six of 43 unrelated male patients had duplication in their dystrophin gene (Table 6 and Fig. 1). In one case, deletion of exon 18 was observed, which was confirmed by PCR (Fig. 2). According to the reference sequence in the LOVD database, the HGVS notation of this deletion is ex18del -> c.2169-?_2292+?del. As a result, deletion of this exon leads to an out-of-frame mutation and, consequently, severe phenotype (www.dmd.nl/). In another case, the ratio of the peak area for exon 58 was 0.2. This exon was covered in our multiplex PCR sets, nevertheless tested negative for the deletion. Subsequent sequencing revealed that a point mutation 8608C>T (R2870X) has occurred within that exon next to the ligation site. The general expectation is that mutations associated with the DMD phenotype produce premature protein truncation or instability of the dystrophin proteins. This nonsense mutation produces a premature termination codon at c.8610 in exon 58. The dystrophin isoforms that are affected by this mutation are Dp427m, Dp260, Dp140pc, and Dp116 (Taylor et al., 2010).
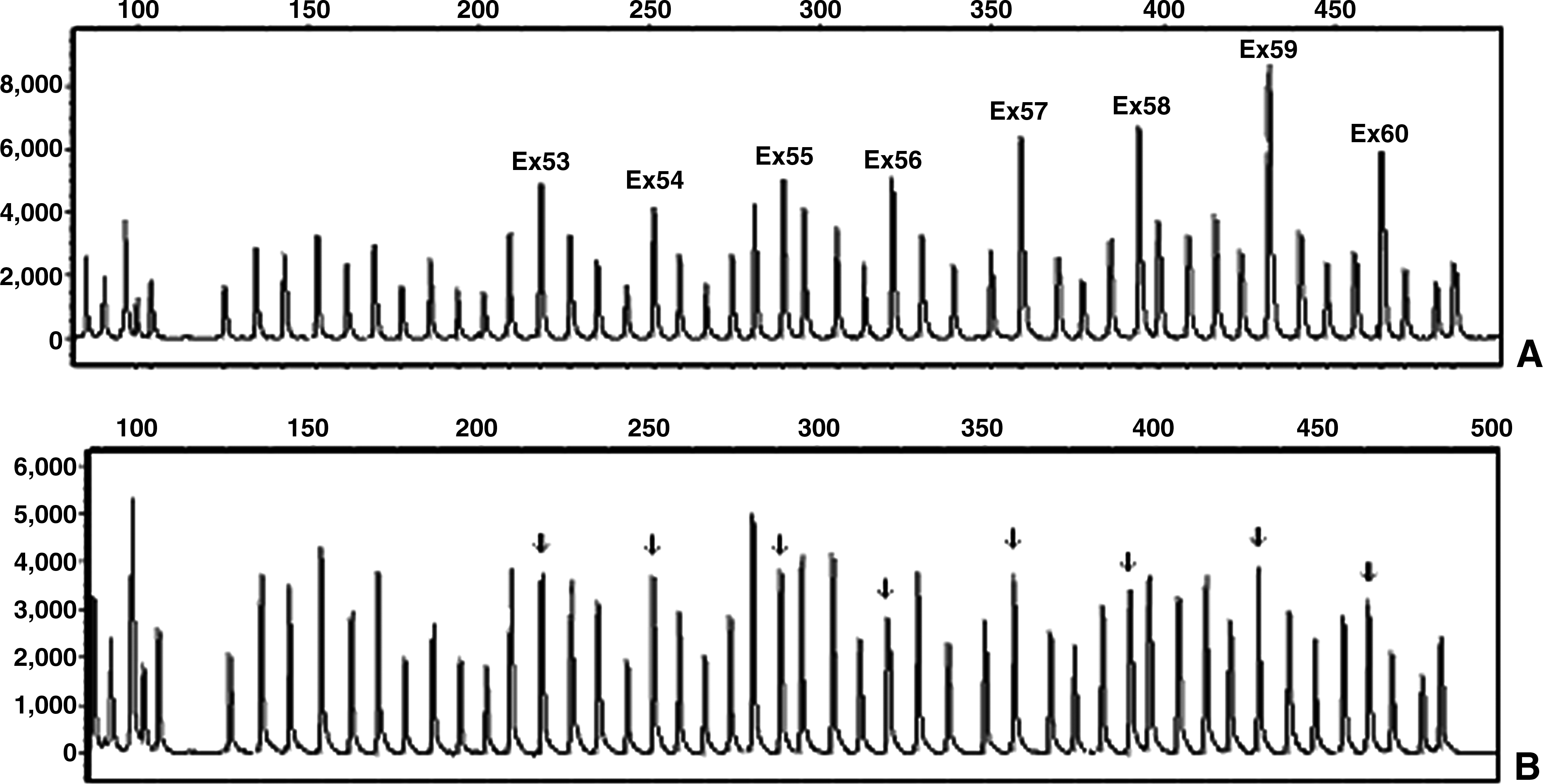
Duplication of exons 53-60 of dystrophin gene in patient 6
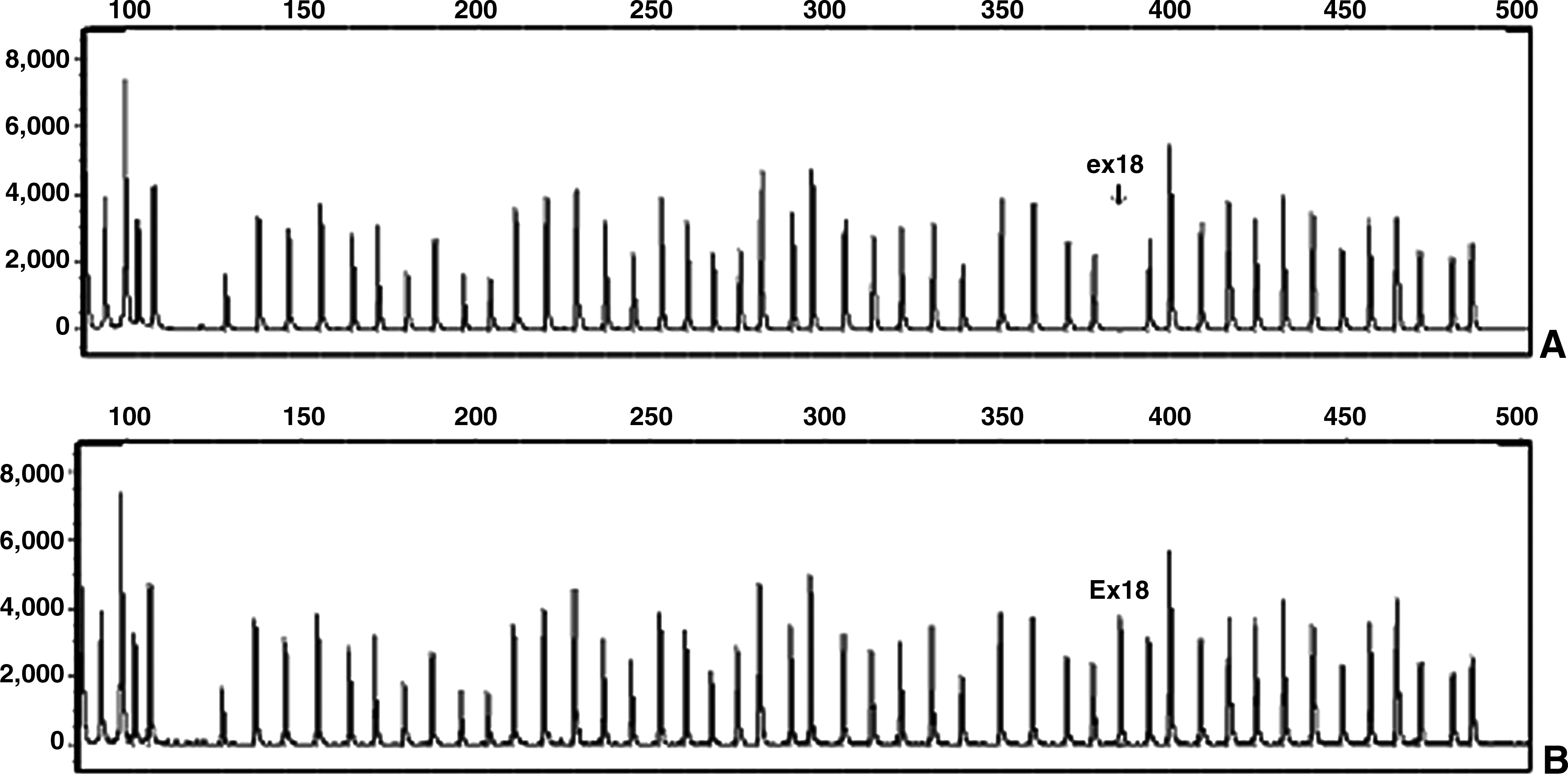
Deletion of exon 18 in patient 7
In females, one person was a carrier of a deletion of exons 49-50 (Fig. 3).
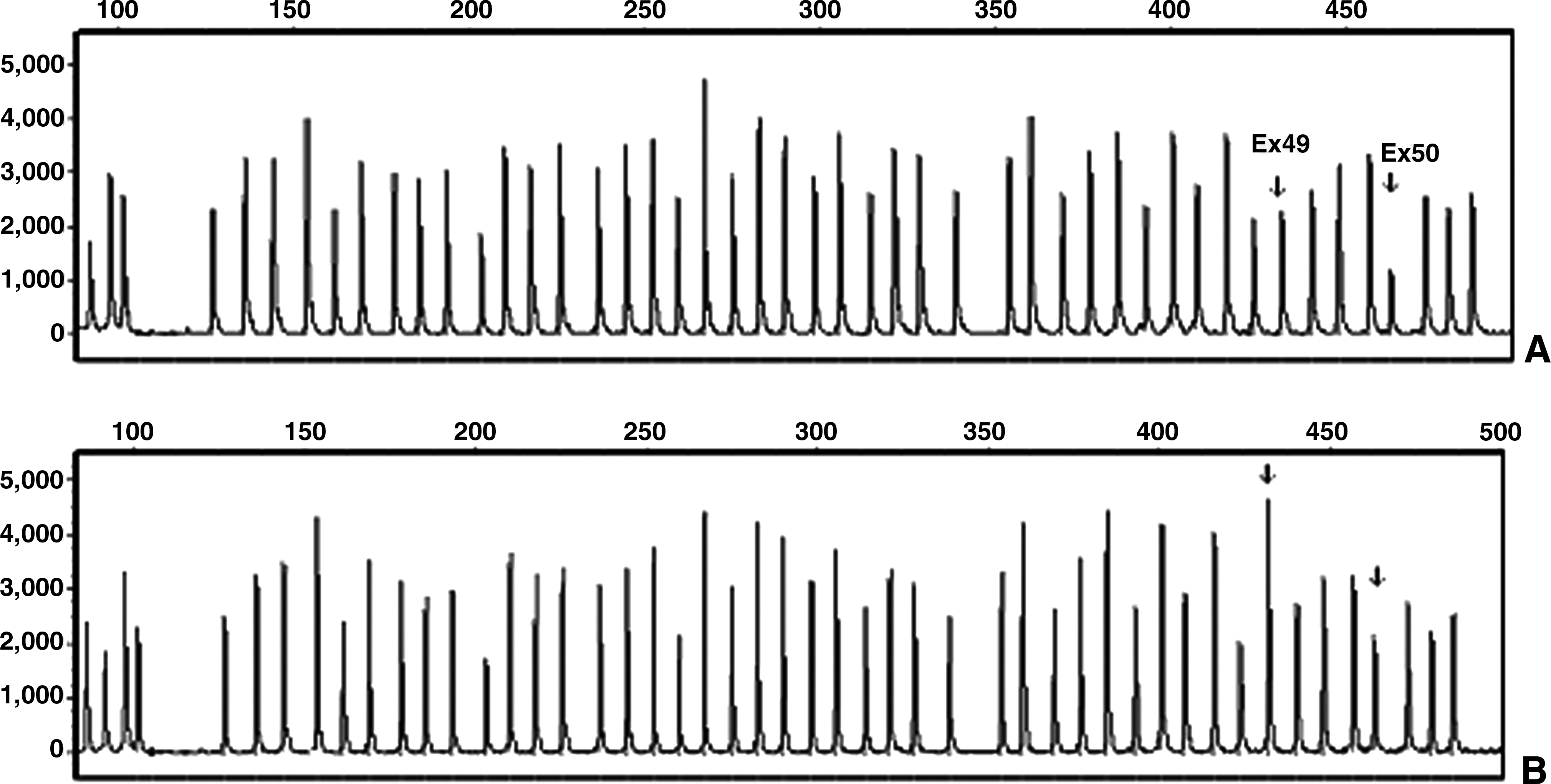
Deletion of exons 49-50 in a carrier female
Discussion
In the majority of DMD/BMD patients, the disease is due to deletions and duplications in the dystrophin gene (Traverso et al., 2006). To choose an appropriate treatment for the patients, knowing the exact mutation is important. Being the largest gene, the rate of recombination is very high in the dystrophin gene and the mutations may be inherited from carrier females or occur de novo. So carrier detection and prenatal diagnosis is crucial. MLPA is a novel technique that has advantages in comparison to other methods. As two sets of MLPA probes are designed for all 79 exons of the dystrophin gene, running just two MLPA reactions is enough to detect all deletions and duplications in this gene, reducing cost and time. Also, using only one fluorescent-labeled primer pair is another advantage of this method (Schouten et al., 2002; Lalic et al., 2005). In contrast to MLPA, multiplex PCR uses different primer pairs for different exons. As these primer pairs have different amplification efficiency during PCR, it is not possible to run just one multiplex PCR for studying all exons. Further, the MLPA method makes it possible to detect duplications in males, as well as deletions and duplications in females, which are not possible by multiplex PCR approach (Schouten et al., 2002). Therefore, MLPA is considered superior to multiplex PCR.
In comparison to FISH, which is limited in detection of multiple loci, MLPA has the advantage of being a multiplex technique. Also, in MLPA, very small (50-70 nt) sequences are targeted, enabling it to identify the aberrations that are too small to be detected by FISH (Schouten et al., 2002).
Finally, compared with array CGH, MLPA is a low-cost, technically uncomplicated method (Schouten et al., 2002).
We performed multiplex PCR on 100 male patients. In 52 patients, the deletions were identified by this approach. DNA samples from 5 of 48 unidentified patients were not available. Therefore, only 43 of them were studied by the MLPA method. In addition, MLPA was performed on 8 male patients with known deletions as positive control samples and 12 females for duplication/deletion characterization. We modified the recommended protocol by performing the ethanol precipitation on the DNA samples. MLPA not only was able to identify all known deletions, but it also could determine the exact size of the deletions in our positive control samples. It revealed duplications and deletions in eight cases that were previously tested negative by multiplex PCR, and interestingly, in one of them the apparent deletion was due to a point mutation. This point mutation, which was previously reported, causes a premature termination codon in exon 58, which is pathogenic (Taylor et al., 2010). In another case, deletion of exon 18 led to an out-of-frame mutation and, therefore, resulted in severe phenotype. Also, one female with heterozygous deletion of exons 49 and 50 was detected.
As the primers in the multiplex PCR method are designed for deletion hotspot regions, deletion in only 1 of 95 patients was missed by this approach. It means that it was able to detect 52 of 53 (98.11%) deletions. However, duplications and point mutations as well as deletions out of hotspot regions are not detected by this method.
According to the literature, partial deletions and duplications of the dystrophin gene account for 60%-70% of the gene defect in the DMD and BMD patients (Den Dunnen et al., 1989; Koenig et al., 1987; Forrest et al., 1988). In our study, in total, 53 of 95 patients (55.78%) had deletion in their dystrophin genes and duplication was identified in 6 patients (6.31%). Therefore, 62.09% of these 95 patients had deletion and duplication in their genes. We could not find any mutations in 35 of 95 patients (36.8%). In these cases, the disease may be due to point mutations, deletions, or insertions of a few nucleotides in the dystrophin gene or the diagnosis may be incorrect. For example, mutation in the FKRP gene results in LMGD 2I, in which the clinical features overlap with DMD (Schwartz and Dunø, 2004).
In conclusion, we found the MLPA method to be rapid, easy to perform, and reliable. The results are highly reproducible on good-quality DNA samples. Also, it is cost effective compared with some other methods such as the quantitative PCR approach. Further, as the instruments required for performing the MLPA method are just commonly used apparatus such as heated lid thermocycler and capillary sequencer, it is possible to perform it in a standard diagnostic laboratory. Moreover, its ability to study all exons of the dystrophin gene and its sensitivity and specificity to determine the exact proximal and distal borders of deletions and duplications in males and females provide a better diagnosis than most other methods. However, confirmation of the results of MLPA is required in cases wherein deletion or duplication of just a single exon is identified. Another disadvantage of the MLPA method is that it misses partial deletions and duplications of the regions for which the probes are not designed. Also, it cannot detect point mutations unless they are near the ligation site of the right and left oligonucleotide probes.
Footnotes
Acknowledgments
The authors thank the patients and their families for their collaboration and the neurologist clinicians who referred them for this study. The personnel of Tehran Medical Genetics Laboratory, especially Dr. Zare and Dr. Karimipoor, are acknowledged for their help and support. The authors are grateful to Dr. Jan Schouten, Dr. Jelger van der Meer, and Eline Sepers from MRC-Holland and highly appreciate their support and technical advice.
Disclosure Statement
No competing financial interests exist.