
Abstract
Genetic factors play a significant role in the etiology of intellectual disability (ID). The goal of this study was to identify microscopically visible chromosomal abnormalities in an Indonesian ID population and to determine their frequency, pattern, and clinical features. A total of 527 intellectually disabled individuals from special schools and institutions in 4 different areas on Java Island, Indonesia, were screened for cytogenetic abnormalities. Additional analyses were carried out for verification or further characterization by using fluorescence in situ hybridization, multiplex ligation-dependent probe amplification, or analysis of the FMR1 promoter CGG(n) repeat. Of the 527 individuals with ID, chromosomal abnormalities were found in 87 (16.5%). Trisomy 21 was the major chromosomal abnormality, identified in 74 patients (14%). Other chromosome abnormalities included 8 X-chromosomal and 5 autosomal aberrations. Details on chromosome aberrations and confirmation analyses are discussed. This study shows that chromosomal abnormalities are an important cause of ID in Indonesia. Cytogenetic analysis is important for an adequate diagnosis in patients and subsequent genetic counseling for their families, especially in developing countries with limited facilities, such as Indonesia.
I
Known causes of ID are biochemical and metabolic defects, chromosomal abnormalities, mutations in single genes (Mendelian disorders and mitochondrial disorders), multifactorial disorders with a polygenic predisposition, and nongenetic causes (Chiurazzi and Oostra, 2000; van Karnebeek et al., 2005). Pathogenic chromosomal abnormalities are the most common genetic cause of ID (Stevenson et al., 2003; Mefford, 2009). Microscopically visible numeric and structural abnormalities account for 7-56% of cases depending on techniques used and patient selection (Fryns et al., 1986; Dereymaeker et al., 1988; Fryns et al., 1990; Felix et al., 1998; Santos et al., 2000; van Karnebeek et al., 2002; Shiue et al., 2004; Dayakar et al., 2010). Down syndrome is the most common chromosomal abnormality causing ID, and it can be easily detected by using routine chromosomal analysis (Tolmie and MacFayden, 2007).
To date, there are few data on the incidence and cause of ID in Indonesia, even though approximately 66,500 pupils have been registered in special schools for intellectually disabled individuals (Kemendiknas 2010). This number, however, is far lower than the total number of ID individuals in Indonesia.
Cytogenetic analysis has not been recognized as a routine diagnostic tool for patients with ID in Indonesia, although the technique is available. Furthermore, genetic disorders have not received much attention from the government and medical practitioners, partly because the main health problems for childhood morbidity and mortality are socioeconomic and environmental, such as malnutrition and infection.
Previous studies in the Indonesian ID population primarily focused on the fragile X syndrome (Hussein, 1998; Faradz et al., 1999). Therefore, this study aimed to determine the prevalence and pattern of microscopically visible chromosomal abnormalities and the clinical features of positive cases in ID individuals in Indonesia.
Materials and Methods
Patient selection and setting
A total of 527 participants (329 males and 198 females) were included in the study. Their ages ranged from 6 to 25 years, and they were from 4 different places on Java Island, Indonesia (Semarang, Temanggung, Yogyakarta, and Bandung). Of the 527 patients, 156 were institutionalized and 371 attended special schools. The majority of the individuals (n=345) appeared to be mildly intellectually disabled, 161 were moderately disabled, and 21 were severely disabled (Table 1). Informed consent was obtained from the parents or legal representatives, and the study was approved by the Ethical Board of the University of Diponegoro/Kariadi Hospital Semarang, Indonesia. All participants underwent a standardized clinical examination before blood was drawn. This examination comprised physical measurements and dysmorphologic assessment.
ID, intellectual disability.
Peripheral blood samples were collected from December 2006 to November 2008, and cytogenetic analysis was performed on all 527 samples. Structural abnormalities were confirmed by multiplex ligation-dependent probe amplification (MLPA) or fluorescence in situ hybridization (FISH). Southern blot analysis was carried out to confirm the presence of cytogenetically visible fragile sites on the X chromosome.
Chromosome cultures and preparations were carried out as described elsewhere (Blennow 2005). One hundred metaphases were screened for fragile sites on each sample. Subsequently, chromosome analysis was performed by using G-banding technique on the level of 400-600 bands. At least 20 metaphases were scored for each patient and karyotyped. If a mosaicism was suspected, 50-100 cells were counted.
FISH analysis was performed by using commercially available probes (Vysis, Inc., Downers Grove, IL) according to standard protocols as previously described (de Bruijn et al., 2001). Genomic DNA of each patient was isolated by using the salting-out method (Miller et al., 1988). MLPA analysis was performed as described elsewhere (Schouten et al., 2002; Koolen et al., 2004). Several probe kits from MRC-Holland (Amsterdam, the Netherlands) were used in these experiments: SALSA P036D and SALSA P070 (probes specifically designed for subtelomeric chromosomal imbalances), SALSA P096 (probes for several ID syndromes), and SALSA P028 (methylation-specific probes for chromosome 15).
Southern blot analysis of the FMR1 CGG(n) repeat was performed as described previously (Oostra et al., 1993; Smits et al., 1994).
Results
Chromosomal abnormalities were found in 87 (16.5%) of the 527 intellectually disabled individuals. Trisomy 21 was the major chromosomal abnormality, occurring in 74 cases (14%). The latter cases consisted of 71 with full-blown classical trisomy 21 (43 males and 28 females), 2 with a mosaicism of trisomy 21 [47,XX,+21(73)/46,XX(27) and 47,XY+21(65)/46,XY(35), respectively] (Table 2), and 1 with a Robertsonian translocation (46,XX,der(14;21)(q10;q10),+21) (Table 3, case 1). The latter patient's mother's karyotype was normal, and her father's sample was not available. Therefore, we could not determine whether this translocation was de novo or inherited from her father.
FISH, fluorescent in situ hybridization; MLPA, multiplex ligation-dependent probe amplification; NT, not tested; SB=Southern blot analysis.
In 13 cases, chromosomal abnormalities other than Down syndrome were detected. Two participants had X-chromosomal aneuploidies (45,X(10)/46,XX(90) and 47,XXX; Table 2). For both females the chromosomal aberration detected is not a satisfactory explanation for their moderate ID. The other 11 cases showed structural chromosome aberrations (cases 2-12, Table 3).
Apart from the t(14;21) case, autosomal structural abnormalities were found in 5 cases (1.0%): 2 unbalanced translocations, 1 balanced translocation, 1 deletion, and 1 isodicentric chromosome. No further confirmation test was performed on the Down syndrome cases, the cases with an X-chromosomal aneuploidy (Table 2), or a case with a large visible terminal Xq deletion (case 7). Five samples from 4 males and 1 female patient were identified to have a fragile site at Xq27.3 (cases 8-12). Southern blot analysis confirmed the presence of a fully methylated expansion (>200 CGG repeats) in the promoter region of the FMR1 gene in each of the 5 cases.
MLPA or FISH analysis was used to confirm the structural chromosomal abnormalities in cases with autosomal aberrations (cases 2-6). Whole chromosome paints of chromosome 18 confirmed a missing part of chromosome 18 in the sample of the patient with 46,XX,del(18)(q21.3→qter)dn (case 2). Further analysis using an 18q telomere FISH probe detected only 1 signal from chromosome 18. Her parent's karyotypes were normal, confirming de novo occurrence. This patient had clinical features resembling those of previously described patients with a similar chromosomal aberration (Kimpen et al., 1991; Kline et al., 1993) (Fig. 1).
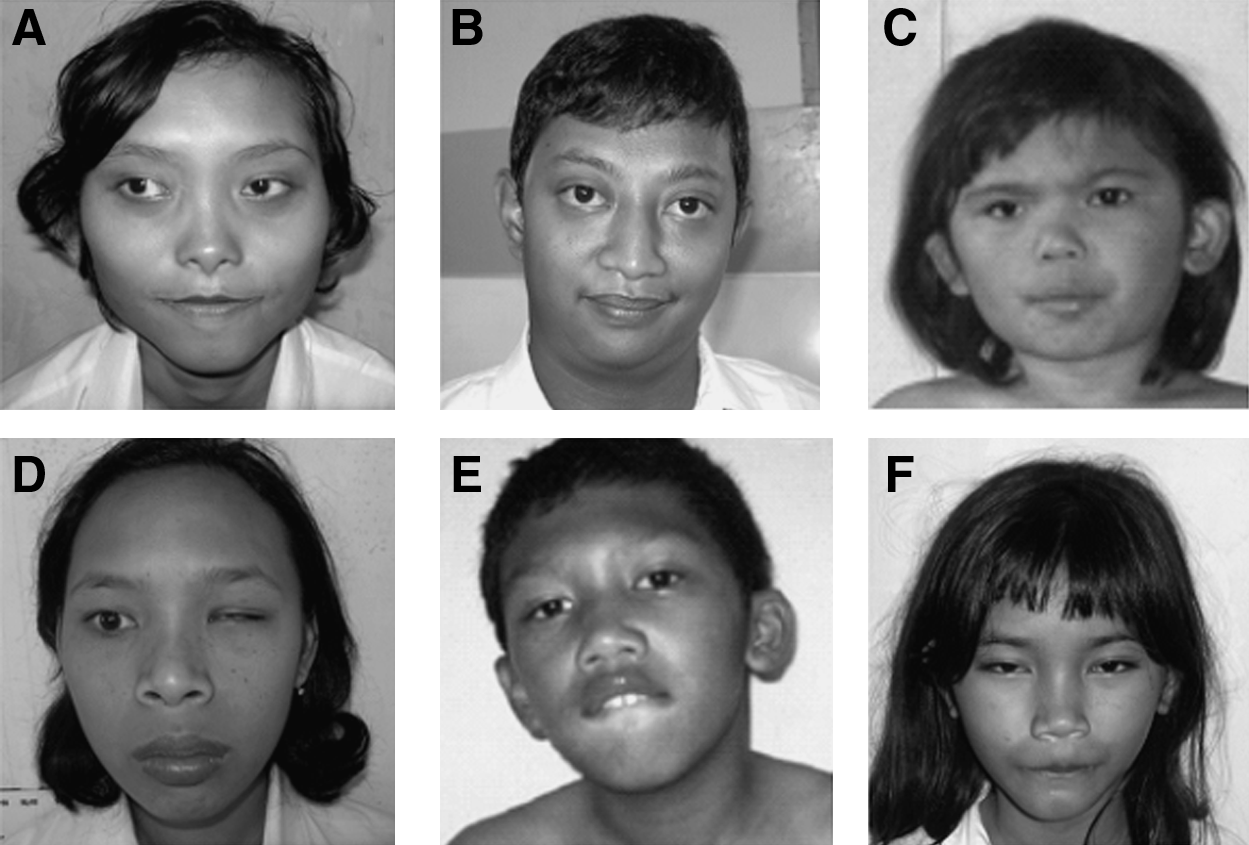
Patients with a chromosomal abnormality.
In case 3, cytogenetic analysis revealed 46,XY,del(4)(p16). However, confirmation with MLPA analysis demonstrated not only a deletion of chromosome 4pter but also a duplication of 8pter (SALSA MLPA kits P036D and P070). Afterwards, FISH was performed by using probes for the subtelomeric regions of chromosome 4p and 8p; indeed, only 1 signal for 4pter and 3 signals for 8pter (1 of which was on the derivative chromosome 4) were detected. Both parents showed normal karyotypes, and carriership of a balanced translocation has been excluded. Therefore, the karyotype of the patient should be designated as 46,XY,der(4)t(4:8)(p16;p23)dn. Further characterization was performed with MLPA analysis by using several probes from the Wolf-Hirschhorn syndrome critical region (WHSCR) (SALSA MLPA kit P096). This analysis revealed a deletion of the entire WHSCR. The cytogenetic and molecular analyses confirmed the clinical diagnosis of Wolf-Hirschhorn syndrome (Fig. 1).
In case 4, cytogenetic analysis revealed a karyotype of 46,XX,add(10)(q26). However, MLPA demonstrated a deletion of chromosome 10qter and a duplication of 4pter. Subsequently, FISH was performed by using probes for the subtelomeric regions of chromosome 10q and 4p. Only 1 signal for 10qter and 3 signals for 4pter (1 of which was on the aberrant chromosome 10q) were detected. Consequently, the karyotype of the patient should be designated as: 46,XX,der(10)t(4:10)(p16;q26). Unfortunately, this patient's parents were unavailable for testing. Because 4pter duplications are reported in patients with and without ID and distinctive facial features (Gerard-Blanluet et al., 2004; Rodriguez et al., 2007), it is suggested that the phenotype in case 4 most likely is due to the deletion of 10qter. The clinical features of this patient are in concordance with the consistent phenotype of patients with a 10q26.1qter deletion, as described by de Vries et al. (2003) (Fig. 1).
In case 5 (Fig. 1), in which chromosome analysis revealed a 46,XX,t(3;12)(p14.1;q21.2) karyotype, further confirmation using MLPA showed a normal result. It is therefore suggested that the aberration was a (cytogenetically) balanced translocation. Neither parent was available for testing. In case 6 cytogenetic analysis showed a 47,XY,idic(15)(q13). MLPA analysis of probes in the 15q11.2-15q15.1 region (MRC Holland kit P028) showed 4 copies of the probes between BP1 and BP4 (including TUBGCP5 and TJP1) and 3 copies of the probes in the TRPM1, KLF13, and CHRNA7 genes (between BP4 and BP5) (Miller et al., 2009). The methylation-specific analysis indicated that the marker was of maternal origin. The patient's parents were unavailable for testing. Clinical features were severe ID, epilepsy, and very poor language expression, which are in fact the main features of isodicentric (15) syndrome (Battaglia 2008) (Fig. 1).
Case 7 had a deletion of part of the long arm of 1 of her X chromosomes [46,X,del(X)(q21→qter)]. She had mild ID and obvious dysmorphisms (Fig. 1). Females with a similar aberration have been reported to show mostly only mild Turner stigmata or subtle dysmorphisms, next to ovarian failure. ID, however, was not reported in these females (Graham et al., 2007), which makes it unlikely that the chromosomal aberration directly caused the ID in this patient. Further investigations, such as X inactivation studies, are needed.
Four males and 1 female expressed a fragile site on the X chromosome. Southern blot analysis confirmed that they all had fragile X syndrome. Three cases (cases 8, 9, and 10) had a full mutation of FMR1, and 2 (cases 11 and 12) showed a mosaicism (premutation and full mutation).
After exclusion of individuals with chromosomal aberrations other than fragile X syndrome (82 patients for the whole population and 46 for the male population), the prevalence of fragile X syndrome in this study is 1.1% (5 of 445) among the whole study population and 1.4% (4 of 283) in the male population.
Discussion
The overall frequency of microscopically visible chromosomal aberrations in this study was 16.5%. This is similar to the rate reported in other studies (13.3%-17.6%) (Fryns et al., 1986; Dereymaeker et al., 1988; Fryns et al., 1990), although different frequencies were found in other studies: 7.9% (van Karnebeek et al., 2002), 22.43% (Shiue et al., 2004), 28.6% (Santos et al., 2000), 34.2% (Felix et al., 1998), and 56% (Dayakar et al., 2010). These differences might be due to variations in inclusion criteria of patients. van Karnebeek and colleagues found a lower frequency of microscopically visible aberrations, possibly because the study was performed in a tertiary care center (outpatient clinic) (van Karnebeek et al., 2002). Some studies generated higher frequencies than our study (Table 4), and this may have occurred because more patients with moderate and severe or profound ID were included. These differences might also be due to preselection of cases without known nonchromosomal causes of ID or multiple congenital anomalies, as was done by Dayakar et al. (2010).
Data are expressed as % (n/n).
The male-to-female ratio in our study was 1.66:1, which is higher than in some previous reports (1.2:1-1.4:1) (Roeleveld et al., 1997; Partington et al., 2000; Macayran et al., 2006; Lin, 2009) but lower than in other studies, which reported a ratio as high as 3:1 (Shin and Lee 1999; Tang et al., 2008). The sex ratio differences might be explained by the differences in selection and case ascertainment (Roeleveld et al., 1997). Another ascertainment bias in our setting might be that parents seek assistance more frequently for boys than for girls because of generally higher expectations for male children.
Fourteen percent of the intellectually disabled individuals in this study had Down syndrome. This finding confirms that the syndrome is the most common chromosomal abnormality involved in ID. Severe cases, such as trisomy 13 and trisomy 18, were not found in our study, most likely because these patients died before they reached school age. The prevalence of Down syndrome in our study is similar to that in previous studies conducted in the Indonesian population(12-14%) (Hussein 1998). In addition, the prevalence of Down syndrome in our study resembled the frequency of 13-15% reported in a white population (Matilainen et al., 1995; van Buggenhout et al., 1999). We found a male-to-female ratio of 1.5:1 in Down syndrome cases, which reflects the male excess in our study population. The proportion of patients with Down syndrome among all male (43 of 329 [13%]) and all female (28 of 198 [14%]) participants, however, was similar, which corresponds to previous reports in this population (Hussein 1998).
The prevalence for fragile X syndrome in this study was 1.1% (5 of 445) among the whole study population and 1.4% (4 of 283) in the male population. A previous study among intellectually disabled individuals in Indonesia that used molecular analysis showed a similar prevalence of 1.9% (5 of 262) in the male population (Faradz et al., 1999). It is also similar to that reported in some studies of white populations (2-3%) (de Vries et al., 1997; Hecimovic et al., 2002; Biancalana et al., 2004). The prevalence could have been higher if molecular analysis had been performed in all 527 patient samples because not all carriers of the FRAXA mutation express the fragile site on karyotyping (Pembrey et al., 2001). However, because of limited availability of molecular testing in Indonesia, cytogenetic studies for fragile X are still a useful tool to detect fragile X(A) and other fragile site abnormalities, including FRAXE, FRAXF, and fragile sites in autosomes (Hussein 1998).
Our study shows that cytogenetic analysis is still a powerful tool to detect genetic abnormalities in the ID population. The fact that cytogenetic analysis can now be performed in Indonesia should be considered by granting agents, such as government and nonprofit organizations, so that they may financially support genetic studies in developing countries such as Indonesia. Furthermore, because common infectious diseases and nutritional problems are becoming less prevalent in Indonesia, diagnostic facilities for genetic diseases must receive a higher priority. Such efforts would extend genetic analysis to more diverse populations than normally studied (Bustamante et al., 2011).
Conclusions
Chromosomal abnormalities play an important causative role in ID in Indonesia. However, because cytogenetic analysis is still not commonly performed in intellectually disabled individuals in Indonesia, the implementation of this technique in a routine diagnostic setting will help to establish a genetic diagnosis in the local setting and will improve the possibilities for genetic counseling to the families.
Footnotes
Acknowledgments
The authors thank Desy Armalina, MD, for her involvement in the clinical examination of patients with Down syndrome. The authors also thank laboratory staff at the Department of Human Genetics RUNMC, the Netherlands, and CEBIOR FMDU, Semarang, Indonesia. This research was funded by the RISBIN-IPTEKDOK 2007/2008 program of the Department of Health, Republic of Indonesia; Excellent Scholarship (Beasiswa Unggulan), Department of National Education, Republic of Indonesia; and the fellowship program of the RUNMC.
Disclosure Statement
No competing financial interests exist.