
Abstract
Background: Hereditary colorectal cancer accounts for approximately 4-5% of all colorectal cancers. The causative genes for familial adenomatous polyposis and hereditary nonpolyposis colorectal cancer are large, making comprehensive analyses difficult. Therefore, high-throughput and practical methods are required to make an early diagnosis of hereditary colorectal cancers and identify high-risk individuals. For this purpose, we developed a novel gene scanning method by high-resolution melting (HRM) analysis. Methods: High-resolution melting (HRM) analysis is a promising prescreening method for nucleic acid sequence variants because of its high sensitivity and high-throughput capability. We evaluated HRM for screening APC, MLH1, MSH2, and MSH6 genes for point mutations, small deletions, and insertions. Simultaneously, we evaluated quantitative polymerase chain reaction-HRM (qPCR-HRM) for screening the MSH2 gene for large rearrangements. Results: All 28 point mutations and 1 large rearrangement were successfully detected by qPCR-HRM analysis. Conclusions: A fast and reliable mutation detection strategy with HRM and qPCR-HRM was used to diagnose hereditary colorectal cancers. Because this method is simple and economical, it may be useful in diagnostic laboratories.
Introduction
C
FAP is an autosomal-dominantly inherited colon cancer characterized by the presence of hundreds to thousands of polyps throughout the colon and rectum. It is caused by germline mutations in the adenomatous polyposis coli APC (MIM# 175100) tumor suppressor gene. Polyposis begins at a mean age of 16 years (range, 7-36 years). Without colectomy, patients with FAP inevitably develop colon cancer, usually by age 35-40 years (Castellsagué E, et al., 2008; Gómez- Fernández N, et al., 2009).
NPCC is an autosomal-dominantly inherited cancer-susceptibility syndrome and is caused by germline mutations in the mismatch repair (MMR) genes, primarily MLH1 (MIM# 120436), MSH2 (MIM# 120435), and MSH6 (MIM# 600678). Germline mutations in MLH1 and MSH2 account for 90% of HNPCC cases (Lawes DA, et al., 2005; Montazer Haghighi M, et al., 2009). HNPCC is the most common cause of hereditary colorectal cancer, with an early age of onset and extracolonic carcinomas.
The causative genes for FAP and HNPCC are large, making comprehensive genetic analyses difficult. Therefore, high throughput and practical methods are required to make an early diagnosis of hereditary colorectal cancers and identify high-risk individuals.
First-line exon screening methods, such as denaturing high-performance chromatography (DHPLC) (Holinski-Feder E, et al., 2001), have been developed. However, the DHPLC method has major drawbacks, such as production of chemical waste, high maintenance costs, and requirement of post-polymerase chain reaction (PCR) manipulations. In addition, DHPLC does not allow high-throughput mutation screening because only 1 sample is used per run.
High-resolution melting (HRM) is a mutation scanning technique that monitors the progressive change in fluorescence caused by the release of an intercalating DNA dye from a DNA duplex as it is denatured by marginal increases in temperature. It is an in-tube method requiring the inclusion of a saturating intercalating dye in the PCR mix and addition of a high-resolution melt step after PCR (Bastien, et al., 2008; Krypuy M, et al., 2007). More recently, innovative approaches have been developed. This strategy, known as qPCR-HRM, is based on a real-time PCR assay (gene dosage quantitative analysis) combined with HRM curve analysis of point mutations in a single tube (Rouleau E, et al., 2009; Coulet F, et al,. 2010; Pallares-Ruiz N, et al., 2010).
Here, we demonstrate that qPCR-HRM analysis is useful in identifying point mutations and large genomic rearrangements in the APC, MLH1, MSH2, and MSH6 gene regions of hereditary colon cancers. Furthermore, we describe the implementation and validation of this approach in a clinical laboratory setting.
Materials and Methods
Patient samples
Samples were obtained from 16 patients with histologically confirmed hereditary colorectal cancers (8 patients each with FAP and HNPCC) and 2 normal controls. Patients with FAP had been previously analyzed for point mutations by direct sequencing. Patients with HNPCC had been previously analyzed for point mutations and large rearrangements by multiplex ligation-dependent probe amplification (MLPA), long-range PCR, and direct sequencing.
DNA extraction
Human genomic DNA was prepared from whole-blood samples by using the MagNA Pure Compact (Roche Diagnostics, Penzberg, Germany) according to the manufacturer's standard protocol. DNA concentration was assessed by using a NanoDrop 1000 spectrophotometer (Coleman Technologies, Orlando, FL). DNA working solutions were prepared at concentrations of 3 ng/μL for qPCR-HRM.
Primer design
Primers were designed to flank the coding region and anneal at 60°C by using the LightCycler Probe Design Software 2.0 (Roche Diagnostics) to calculate the “Tm.” All primers were designed to be 100-250 bp in length. All the primers used in this study are listed in Supplementary Table S1 (supplementary data are available online at www.liebertpub.com/gtmb). Two primer pairs were designed for the coding sequence of the ALB and ERBB2 genes as diploidy references (Rouleau E, et al., 2009). Common primers were used for qPCR-HRM and direct sequencing was used to screen for exons containing mutations.
Assay conditions
HRM and qPCR were performed in a single run on a LightCycler 480 (Roche Diagnostics) in a reaction mix containing 9 ng of genomic DNA, 0.2 μM of each primer, and 3 mM MgCl2 in the LightCycler 480 High Resolution Melting Master containing Resolight dye (Roche Diagnostics) with PCR-grade water adjusted to a total volume of 17 μL. The QIAgility (Qiagen, Courtaboeuf, France) automated PCR workstation was used to set up qPCR-HRM samples. The same cycling conditions were used for all reactions; PCR was performed with an initial denaturation step at 95°C for 10 min, followed by 40 cycles of 95°C for 10 s, a touchdown from 64°C to 54°C for 10 s (1°C/cycle), and a final step at 72°C for 10 s. After amplification, products were heated to 95°C for 1 min and cooled to 40°C for 1 min to favor heteroduplex formation. For the melting step, the temperature was raised from 65°C to 95°C with a ramp rate of 0.02°C/s and 25 acquisitions/°C. Each experiment included wild-type DNA. Quantitative PCR analysis was performed to identify large gene rearrangements using LightCycler 480 Relative Quantification Software (ΔΔCt method). HRM curve analysis was performed by using the LightCycler 480 Gene Scanning Software (version 1.5) to detect nucleotide variation.
Direct sequencing
All regions of the APC gene and MLH1, MSH2, and MSH6 gene mutations and genetic variants detected were confirmed by sequencing PCR products. The PCR products generated after HRM can be sequenced directly. Sequence detection was performed by using the Applied Biosystems 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA).
Results
DNA from the 8 patients with FAP was tested for APC gene analysis, and DNA from the 8 patients with HNPCC was tested for MLH1, MSH2, and MSH6 gene analysis. All PCR products obtained after HRM were subjected to electrophoresis in 2% agarose to indicate the presence of unique products of expected sizes (data not shown). For HRM, the maximum of the observed range of normality was between +3 and −3 in the normalized and temperature-shift difference plots for wild-type amplicons. Figure 1 shows the process of rapid screening for gene mutations using HRM.
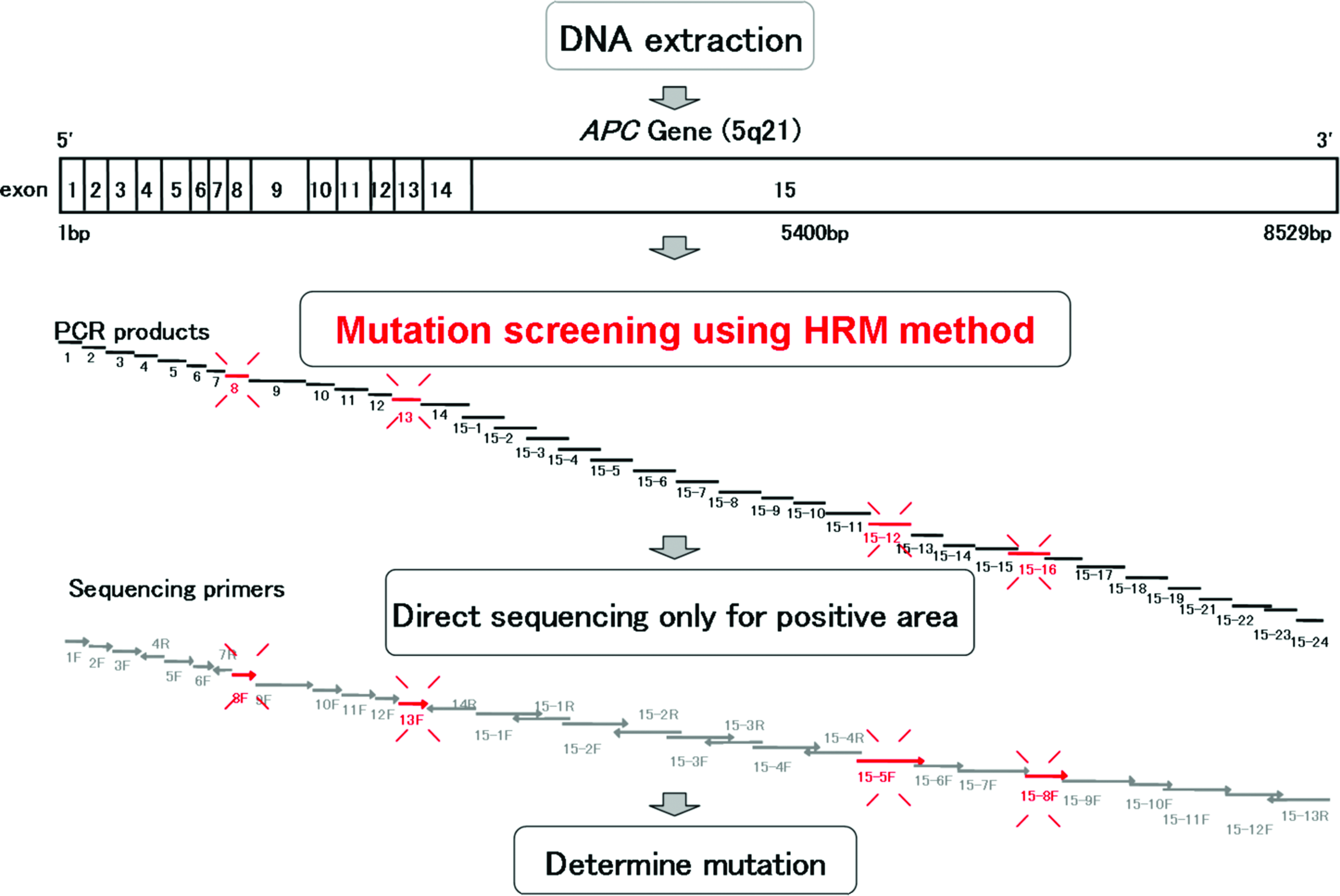
Process for the rapid prescreening of gene mutations using high-resolution melting (HRM). PCR, polymerase chain reaction. Color images available online at www.liebertonline.com/gtmb
All the point mutations detected by direct sequencing were detected by HRM using the LightCycler 480 software (Table 1). We detected 14 different mutations in the APC gene: 4 nonsense mutations, 2 frameshift mutations, 1 missense mutation, and 7 silent mutations. Moreover, we detected 14 different mutations in the MLH1, MSH2, and MSH6 genes: 1 large deletion mutation, 7 missense mutations, 4 base insertion mutations, 1 silent mutation, and 4 intron mutations. No amplicon had false results in any of the samples.
DEL, deleterious mutation; POL, polymorphism; UV, unclassified variant.
Figure 2 illustrates several results: exon 12 of MSH2, missense mutation c.1864 C>T; exon 15 of APC, nonsense mutation c.2507C>G; exon 9 of APC, frameshift mutation c.1100_1101delCT; exon 10 of MSH6, insertion mutation c.4065_4066insTTGA; exon 13 of APC, frameshift mutation c.1634delC and silent mutation c.1635A>G; and exon 10 of MSH2, mutation c.1661 +12G>A. These mutations were clearly detected by the normalized and temperature-shifted difference plot.

Example of different mutations detected by high-resolution melting (HRM). Direct sequencing was used to confirm and characterize the affected exons identified by HRM.
Figure 2F shows HRM analysis of a homozygous mutation. The homozygous mutation sample (105AA) could be distinguished from wild-type (106GG) and heterozygous (104GA) mutation samples. Deletion in exon 1 of MSH2 (approximately 10 kb) was detected by qPCR-HRM (Fig. 3).
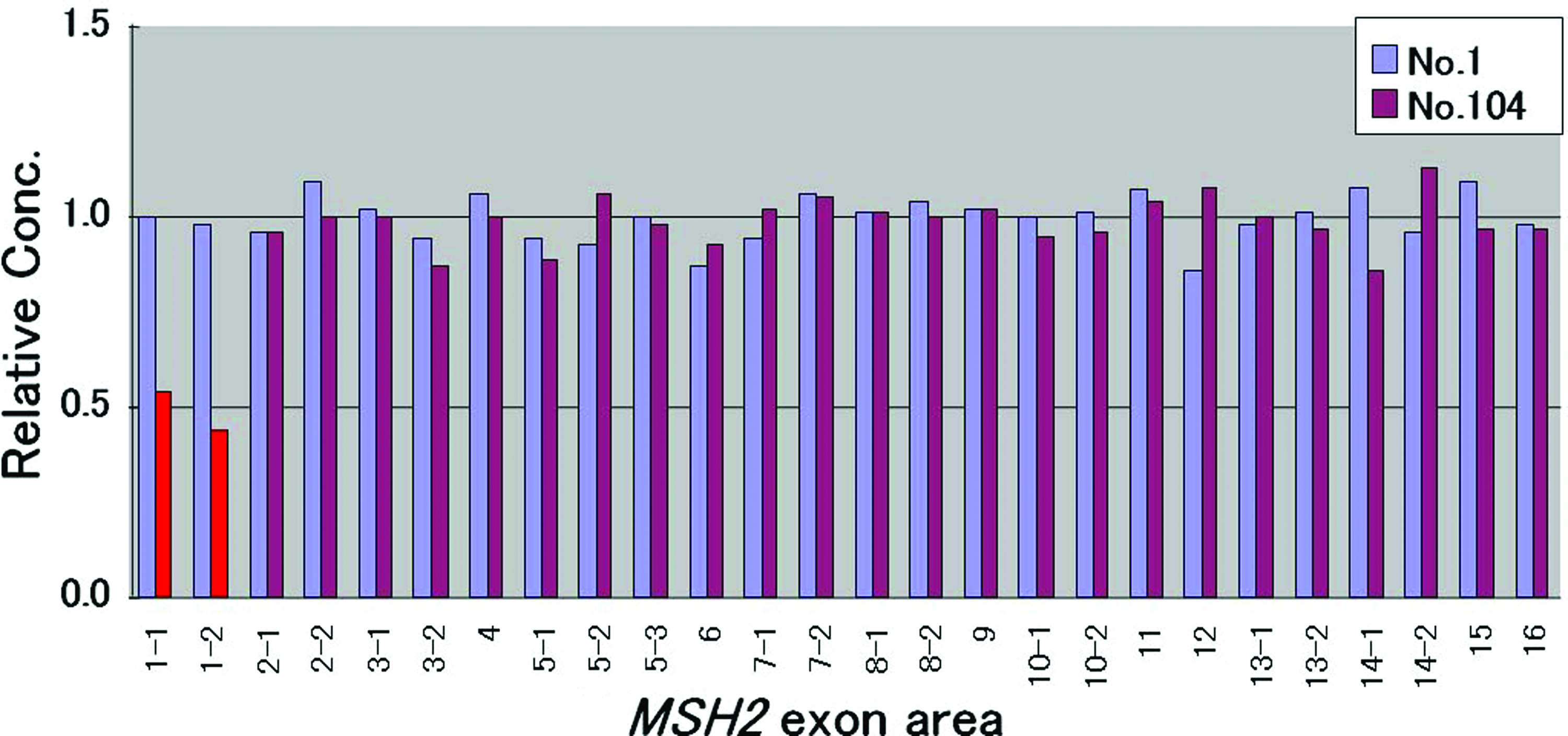
Quantitative polymerase chain reaction analysis for each MSH2 amplicon in normalized Nex 2-ΔΔCt to a reference gene (ALB). The blue columns represent a control (no.1), and the purple columns represent a patient (104); for the latter, the deletion of exon 1 is shown in red. Color images available online at www.liebertonline.com/gtmb
Discussion
Each of the numerous methods used to detect gene mutations has advantages and disadvantages. Prescreening methods such as DHPLC are advantageous because the amount of sequencing required is reduced (Lawes DA, et al., 2005). However, the identification of germline mutations is both labor intensive and expensive. HRM analysis is a new and attractive gene scanning tool that quickly performs PCR and identifies sequence alterations without requiring post-PCR treatment. In this study, we demonstrate the utility of HRM analysis as a reliable and sensitive technology for the detection of gene mutations in hereditary colorectal colon cancer. The aim of this study was to build a simple and quick screening system using the HRM method for mutation screening of APC, MLH1, MSH2, and MSH6 genes involved in hereditary colorectal cancer.
We designed 142 PCR amplicons for HRM analysis, which covered the complete coding region of APC, MLH1, MSH6, and MSH2 genes. All point mutations and rearrangements were successfully detected by qPCR-HRM analysis. We detected 14 different mutations in the APC gene as well as 14 different mutations in the MLH1, MSH2, and MSH6 genes. All the mutations were characterized by direct sequencing (except large deletions). HRM is predicted to have close to 100% sensitivity as each heterozygous mutation should create a readily detectable heteroduplex (Gundry et al., 2008). Different sequence variants can be identified according to differences in melting curves using LightCycler 480 gene scanning software.
Furthermore, we successfully detected large deletions in MSH2 exon 1 by using qPCR-HRM analysis. A major advantage of qPCR-HRM is that post-PCR manipulations, which can increase the risk for error or contamination, are not required. The method also allows detection of point mutations and large rearrangements in 1 step. Importantly, no false-positive or false-negative results were obtained with HRM in the present study, indicating that this high-throughput rapid screening method has very low false-negative and false-positive rates. In addition, the method used in this study can quickly identify genetic variants or mutation even in large genes, such as APC, MLH1, MSH2, and MSH6. However, perhaps the most important advantage of qPCR-HRM is that it can save both time and money.
In summary, we present a fast and reliable mutation detection strategy for hereditary colorectal cancers. We clearly detected all point mutations and large rearrangements. In our opinion, HRM and qPCR-HRM may become methods of choice for hereditary colorectal cancer gene mutation screening in clinical settings. Because this method is simple and economical, it may be useful in diagnostic laboratories.
Footnotes
Acknowledgments
We would like to thank Bahityar Rahmutulla of the Department of Molecular Diagnosis, Graduate School of Medicine, Chiba University, Japan, for helpful discussions and suggestions.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.