
Abstract
The isolation of high-quality RNA and DNA from various specimens is essential to perform reliable molecular diagnostic assays. In routine diagnostics of hematologic malignancies isolation of high-quality RNA is a prerequisite. We used QIAsymphony technology (QST) using a customized RNA CT 800 V6 protocol for automated semi-high-throughput isolation of RNA from human specimens and compared the results for breakpoint cluster region-c-abl oncogene 1 (BCR-ABL1) quantification by real-time quantitative polymerase chain reaction (RQ-PCR) and detection of JAK2 V617F mutations by reverse-transcriptase PCR (RT-PCR) on QST RNA with RNA isolation performed with our routine manual method using RNA-Bee (RB). QST RNA was isolated with and without the addition of β-mercaptoethanol (BME). Addition of BME to the lysis buffer RLT Plus resulted in consistently lower Ct values in analyses of the reference gene porphobilinogen deaminase (PBGD). Further, the BCR-ABL1 mRNA levels of the QST RNA isolation were highly consistent with RB RNA isolation, only when the lysis buffer RLT Plus in addition contained BME. Moreover, cases of myeloproliferative neoplasms (MPN) with low levels of JAK2 V617F mRNA were even missed in QST when lysis buffer RLT Plus was used, but they were readily detected after addition of BME.
Introduction
T
Hematologic malignancies often carry chromosomal translocations (Heim and Mitelman, 2009). These chromosomal fusions occur in intronic regions of genes and result in the expression of fusion transcripts. These fusion mRNA transcripts are the targets in routine molecular diagnostics and thus require RNA isolated from the malignant cells. Standardized high-quality isolation of mRNA from bone marrow and blood samples is therefore essential for reliable routine molecular analyses.
CML is a myeloproliferative clonal disease that is consistently associated with the presence of the reciprocal translocation (9;22)(q34;q11), which juxtaposes the c-ABL (ABL1) oncogene on chromosome 9 to the breakpoint cluster region gene (BCR) on chromosome 22, resulting in a new fusion gene BCR-ABL1. This fusion gene encodes the BCR-ABL1 tyrosine kinase fusion protein with constitutive kinase activity, essential for leukemogenesis. Specific tyrosine kinase inhibitors (TKIs), such as imatinib, nilotinib, and dasatinib, have become first-line treatments in case of CML. Molecular monitoring for CML patients treated with these TKIs is indispensable (Branford et al., 2006; Hughes and Branford 2006; Hughes et al., 2006; Cross et al., 2008).
Essential thrombocytosis (ET), polycythemia vera (PV), and primary myelofibrosis (PMF) are clonal hematopoietic stem cell disorders that share an overproduction of myeloid cells and constitute the BCR-ABL1 negative MPN. The discovery of the JAK2 V617F mutations in these diseases in 2005 has improved diagnostics of these diseases considerably (Baxter et al., 2005; James et al., 2005; Kralovics et al., 2005; Levine et al., 2005). The JAK2 mutation is a single amino acid substitution at position 617, that is, a substitution of valine (V) for phenylalanine (F). As a result of this substitution, JAK2 becomes constitutively activated, independent of ligand. JAK2 V617F mutations are found in approximately 95% of PV and 55% of ET and PMF cases. Reliable analyses of JAK2 V617F mutations, on either RNA or DNA, are required for optimal diagnostics of ET, PV, and PMF. This has led to reclassification of these diseases by the World Health Organization (WHO) (Vardiman et al., 2009).
We applied QIAsymphony technology (QST) using a customized RNA CT 800 V6 protocol for automated semi-high-throughput isolation of RNA from bone marrow and peripheral blood specimens and compared these results with RNA isolation performed with our routine manual method using RNA-Bee (RB). The QST RNA isolation required the addition of β-mercaptoethanol (BME) to the lysis buffer RLT Plus because this addition resulted in consistently lower Ct values in real-time quantitative analyses of the reference gene porphobilinogen deaminase (PBGD). Further, the BCR-ABL1 mRNA levels of the QST RNA isolation were highly consistent with RB RNA isolation, only in case of BME addition to the lysis buffer RLT Plus. Moreover, MPN cases with low levels of JAK2 V617F mRNA were even missed in QST when lysis buffer RLT Plus was used, but they were readily detected after addition of BME to this buffer.
Materials and Methods
Cells and patient samples
White blood cells (WBCs) were isolated from bone marrow or peripheral blood samples of CML patients during treatment or samples of patients with possible ET, PV, or PMF, following density centrifugation (buffy coat) at 300 g at 25°C. Duplicate samples of 107 cells were lysed in 1.0 mL RB or 0.8 mL buffer RLT Plus (Qiagen, Venlo, The Netherlands), the latter either with or without the addition of 1:100 v/v BME (commercially available solutions are usually 14.3 M).
RNA isolation and quality control
RNA was isolated from 107 cells using RB following the protocols of the manufacturer (Bio-Connect BV, Huissen, The Netherlands), with the only exception that the RNA pellet was washed twice with 1 mL 70% ethanol after precipitation with isopropanol.
RNA was isolated by QST using the QIAsymphony RNA kit (cat. no. 931636, Qiagen). In brief, 107 WBCs were disrupted in 800 μL RLT buffer Plus with or without BME. No bead mill, Qiagen TissueRuptor or other rotor-stator homogenizer, syringe or needles were used to homogenize the cells. These samples were processed with a customized RNA CT 800 V6 protocol on the QIAsymphony SP instrument. On our request, Qiagen Application Lab department modified the standard RNA CT-800 V6 protocol steps with the aim to exclude any cross-over contamination from sample to sample. Therefore, the multidispense option was disabled in the latest standard RNA CT 800 V6 protocol so that a new set of tips was used for every dispensation and aspiration step. This modification was registrated by Qiagen as customized application CR2194. Moreover, at the end of the QST RNA isolation the automated transfer of the final eluate was performed by a modified pipetting step directly into 4°C precooled 0.5 mL Amicon Ultra centrifugal filters provided with an Ultracel 30K membrane (cat. no. UFC5030BK; Millipore Ireland Ltd., Cork, Ireland) and registrated as the customized application CR4053. Both customized applications were uploaded into the QIAsymphony instrument as assay control file sets using the supervisor user ID. The final eluates were subsequently concentrated by centrifugation of the Ultra centrifugal filters in a Thermo Scientific, Heraeus Fresco 17 or 21 centrifuge at 14,000 g for 15 min at 4°C. Thirty microliters of the concentrate was immediately transferred into a 4°C precooled 1.5 mL Sarstedt micro tube PP (ref. no. 72.692.100l; Sarstedt, Nümbrecht, Germany), capped and subsequently stored at −80°C.
The quality of the RNA was monitored with gel electrophoresis and RNA concentrations were determined using the NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE).
cDNA synthesis
cDNA was synthesized from 1 μg of RNA using random hexamer priming, essentially as described (Van der Reijden et al., 2001). cDNA prepared from 25 ng of RNA was used for all real-time quantitative polymerase chain reaction (RQ-PCR) and RT-PCR amplifications.
RQ-PCR BCR-ABL1 and PBGD
RQ-PCR amplification for BCR-ABL1 and the reference gene PBGD was performed with the ABI Prism 7500 Real Time PCR system (Life Technologies-Applied Biosystems, Carlsbad, CA) using 25 μL mix containing 125 μM deoxyribonucleoside triphosphates (dNTPs) (Amersham Pharmacia Biotech, Roosendaal, The Netherlands); 7.5 pmol forward and reverse primer (Life Technologies, Carlsbad, CA); 1 mM MgCl2 (BCR-ABL1) or 5 mM MgCl2 (PBGD); 4 pmol probe (BCR-ABL1) or 1,25 pmol probe (PBGD), labeled at the 5′ end with the reporter dye molecule FAM (6-carboxy-fluorescein) and at the 3′end with the quencher dye molecule 6-carboxy-tetramethylrhodamine (TAMRA) for both BCR-ABL1 and PBGD (Eurogentec Nederland BV, Maastricht, The Netherlands); 5 μL 10×buffer A and 1.25 U AmpliTaq Gold (Life Technologies-Applied Biosystems). The following primers and probe were used to determine the BCR-ABL1 expression levels: primer T.BA FOR: 5′-CCGCTGACCATCAATAAG GAA-3′, primer T.BA REV: 5′-TCAGACCCTGAGGCTCAA AGTC-3′, and probe BCR-ABL1 6-FAM 5′-AAGCCCTTC AGCGGCCAGTAGCA-3′ TAMRA. The following primers and probe were used to determine the PBGD reference gene expression: PBGDrev 5′- GGGTACCCACGCGAATCAC -3′, PBGDforw 5′- GGCAATGCGGCTGCAA -3′, and probe PBGD 6-FAM 5′- CATCTTTGGGCTGTTTTCTTCCGCC-3′ TAMRA. The thermal cycling conditions included 10 min at 95°C followed by 45 cycles (BCR-ABL1) or 40 cycles (PBGD) of denaturation for 30 s at 95°C and annealing/extension at 60°C for 60 s.
To quantify the relative expression levels of BCR-ABL1 in the CML samples the Ct values were normalized for the endogenous reference PBGD (ΔCt=Cttarget−CtPBGD) and compared to BCR-ABL1 expression in a calibrator, that is, the BCR-ABL1 positive cell line K562, using the ΔΔ Ct method (ΔΔCt=ΔCtCML sample−ΔCtCalibrator). We used the ΔΔCt value to calculate relative expression (2−ΔΔCt). The sensitivity of this assay monitored by dilution of K562 RNA carrying a BCR-ABL1 fusion transcript into HL60 lacking a BCR-ABL1 fusion transcript. The sensitivity of the assay was consistently 4.5 log, that is, 10−4.5.
RT-PCR JAK2 V617F mutation
Allele specific PCR for JAK2 c.1848_1849TG>CT or JAK2 p.V617F mutation (denoted as JAK2 V617F mutation throughout the article) detection was performed according to Baxter et al. (2005). However, the assay was adapted such that JAK2 V617F mutations were detectable using cDNA as template.
For JAK2 V617F mutation analysis, an allele-specific PCR on each individual sample was performed using the allele-specific forward primer JAK2 FOR 5′-AGCATTTGGTTTTAAATTATGGAGTATATT-3′ in combination with the reverse primer JAK2 REV 5′-GCATGGCCCATGCCAACTG-3′ (165 bp PCR product). All PCR were performed in duplicate in 0.4 mM dNTP, 10 pmol forward and reverse primer, 4 mM MgCl2, Taq polymerase, and 1xTaq polymerase buffer (Life Technologies-Invitrogen, Carlsbad, CA). Cycling conditions were as follows: one cycle for 5′ at 95°C, 32 cycles for 1′ at 94°C, 1′ at 66°C, 1′ at 72°C, and one cycle for 10' at 72°C.
Results
PBGD and BCR-ABL1 RQ-PCR and JAK2 V617F mutation detection show inconsistent results between QST and RB RNA isolation
RNA was isolated from 30 bone marrow biopsies or peripheral blood samples of patients with CML with either the QST or our routine manual RB isolation. Initially, no BME addition was used in the sample preparation for the QST RNA isolation. The overall RNA yield from 107 WBCs was similar for both isolation methods (30 samples in duplicate, RB yield: 7.5μg [SD 2.7] and QST yield: 7.2 μg [SD 2.4]).
Expression levels of the reference gene PBGD were determined using RQ-PCR. The mean PBGD Ct value on RNA isolated with RB was 25.93, and with the QST 27.39, differing 1.46 Ct value. The PBGD Ct values in QST isolated RNA were consistently higher compared with RB isolated RNA. Moreover, the PBGD quantities demonstrated only limited conformity between the QST and RB RNA isolation (Rcorr=0.238, Fig. 1). Since the Ct PBGD values of the QST RNA isolation were consistently higher than the RB RNA isolation and showed limited correlation we decided to change our isolation strategy. Therefore, a relatively small number of samples were analyzed.
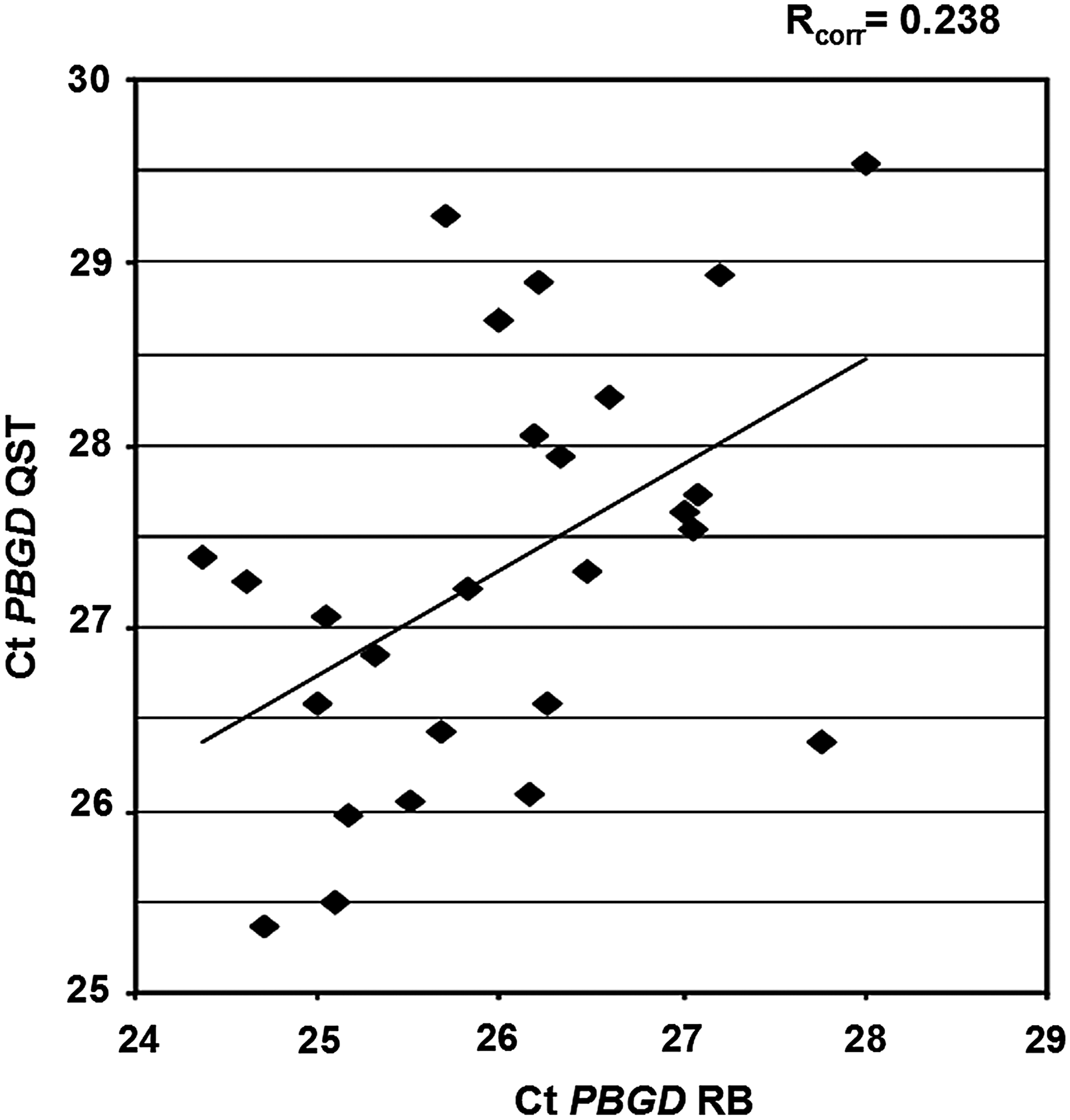
Comparison of real-time quantitative polymerase chain reaction (RQ-PCR) Ct values of the reference gene porphobilinogen deaminase (PBGD) in 30 bone marrow and peripheral blood RNA samples of patients with chronic myeloid leukemia (CML). RNA isolated with QIAsymphony technology (QST) in the absence of β-mercaptoethanol (BME) and RNA isolated with RNA-Bee (RB).
In only 9 out of the 30 samples, expression levels of BCR-ABL1 were determined in RNA isolated with both methods, which precludes reliable conclusions regarding quantification of the BCR-ABL1 levels after PBGD normalization. Of note, the nine samples for which the BCR-ABL1 levels could be quantified showed consistent values between the two RNA isolation methods (R corr=0.995, data not shown). This does demonstrate that even though the PBGD values of the QST RNA isolations show lower conformity with our RB isolations, the BCR-ABL1 after PBGD normalized values are highly comparable.
The JAK2 V617F mutation detection assay was performed on 35 samples of patients with possible ET, PV, or PMF with either the QST or our routine RB RNA isolation. The mean PBGD Ct value on RNA isolated with RB was 26.82, and with the QST 28.07, differing 1.25 Ct value. The PBGD quantities again demonstrated limited conformity between the QST and RB RNA isolation, similar to the samples used for BCR-ABL1 quantifications (Rcorr=0.570, data not shown). More importantly, two out of eight samples that carried high levels of JAK2 V617F mRNA based on the RB RNA isolation demonstrated lower JAK2 V617F mutation signals when QST isolated RNA was used (Fig. 2, patient 1 and 4). Moreover, in a sample with low levels of JAK2 V617F mRNA based on the RB RNA isolation the JAK2 V617F mutation was even missed using the QST RNA (Fig. 2, patient 3).
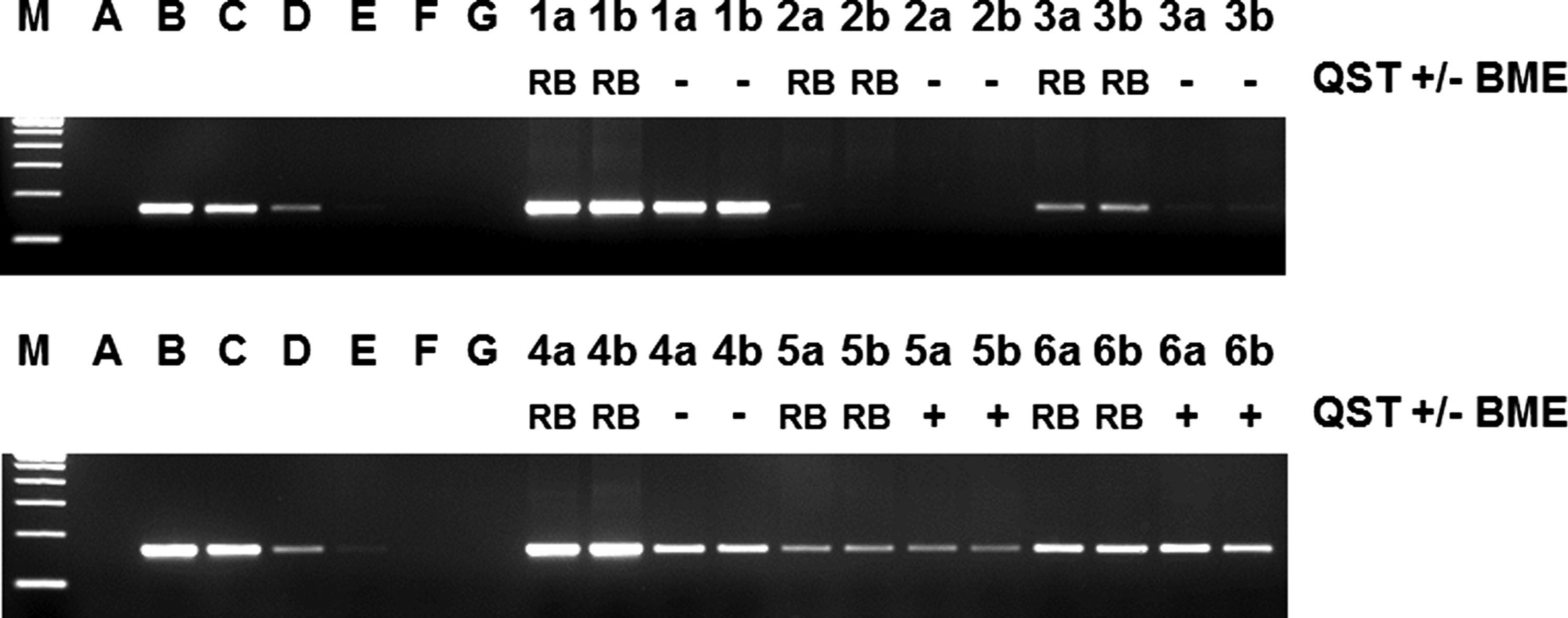
Representative JAK2 V617F detection using an allele-specific reverse-transcriptase-PCR followed by agarose gel electrophoresis on RNA isolated with QST in the absence (−) and presence (+) of BME or RNA isolated with RB. Patient 1+4: JAK2 V617F-positive patient samples; Patient 2: JAK2 V617F-negative patient samples; Patient 3, 5, and 6: JAK2 V617F weak positive patient samples. Most important, patients with low level JAK2 V617F mutation load are clearly detected with the addition of BME (patient 5 and 6), whereas these low levels were barely detectable without the addition of BME (patient 3). A: negative control HL60 RNA; B-G: 1/10 dilution series of the JAK2 V617F containing HEL cell line in the HL60 cell line, undiluted-10−5 dilution; a and b: duplicate analyses on two independent samples of the same patient.
After addition of BME PBGD and BCR-ABL1 RQ-PCR and JAK2 V617 mutation detection show greatly improved results with QST compared to RB RNA isolation
RNA was isolated from 112 bone marrow biopsies or peripheral blood samples of patients with CML with either QST or RB, or with both RNA isolation methods. The QST RNA isolation was carried out with lysis buffer RLT Plus containing BME. The overall RNA yield from 107 WBCs was similar for both isolation methods (112 samples in duplicate, RB yield: 7.5 μg [SD 2.7] and QST yield: 7.4μg [SD 3.1]). Expression levels of the reference gene PBGD were again determined using RQ-PCR. The mean PBGD Ct value on RNA isolated with RB was 26.24, and with the QST 25.82, differing 0.42 Ct value. The PBGD Ct values of QST isolated RNA were consistently lower compared with RB isolated RNA. Moreover, the PBGD quantities demonstrated stronger concordance between the QST and RB isolation (Rcorr=0.639, Fig. 3).

Comparison of RQ-PCR Ct values of the reference gene PBGD in 112 bone marrow and peripheral blood RNA samples of patients with CML. RNA isolated with QST in the presence of BME and RNA isolated with RB.
In 51 out of the 112 samples expression levels of BCR-ABL1 were determined in RNA isolated with both methods. The BCR-ABL1 Ct values (Rcorr=0.953, Fig. 4A) and the quantification of the BCR-ABL1 levels after PBGD normalization (Rcorr=0.967, Fig. 4B) showed very high correlation between QST and RB isolation. Of note, six samples with very low level BCR-ABL1 (<10−4.5) were detectable using RB RNA, and missed with QST RNA, whereas four samples with very low BCR-ABL1 levels were detectable using QST RNA, and missed with RB RNA.
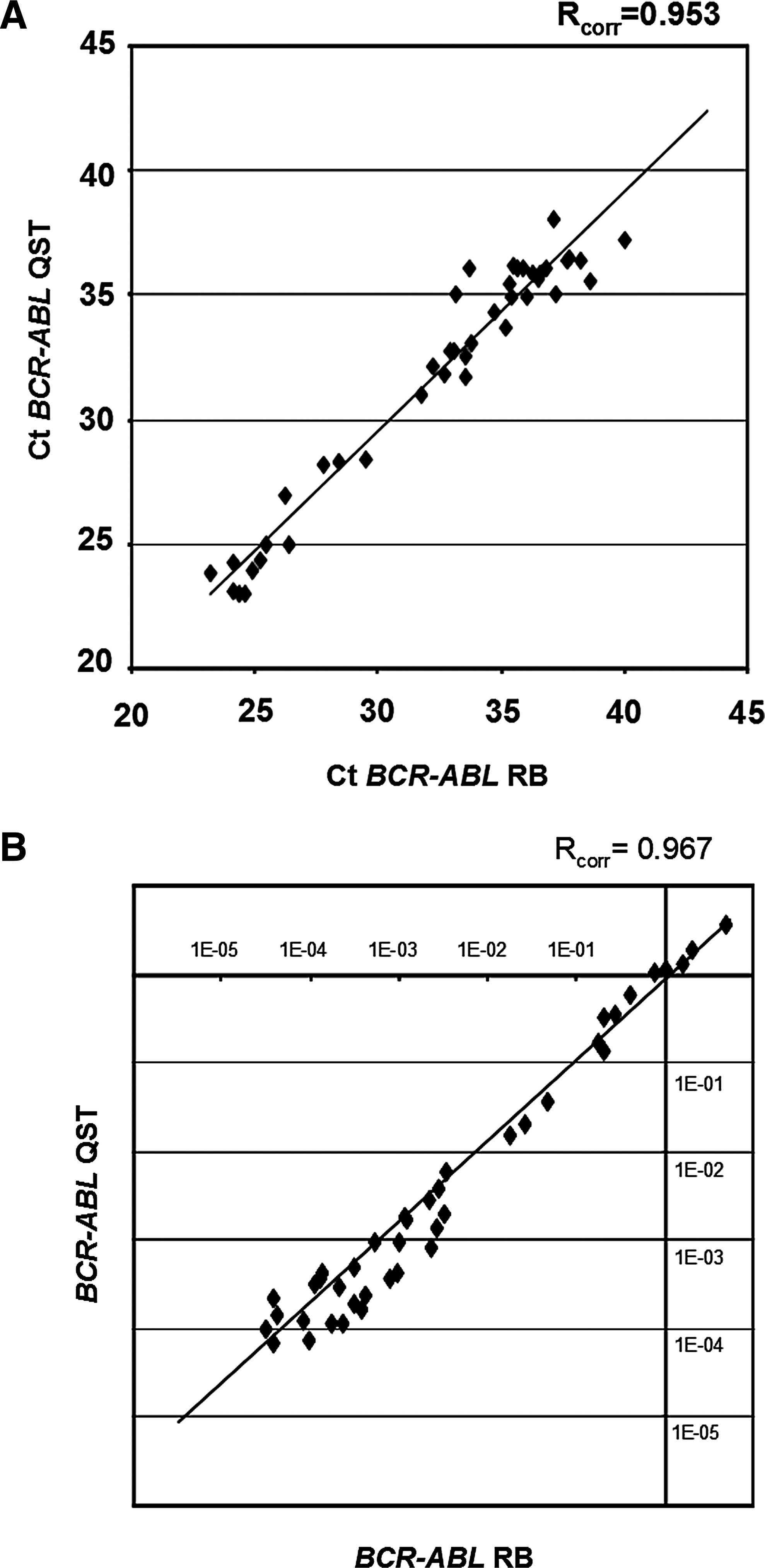
Comparison of RQ-PCR Ct values of the oncogene breakpoint cluster region-c-abl oncogene 1 (BCR-ABL1)
The JAK2 V617F mutation detection assay was performed on 62 RB RNA and QST RNA samples. The overall RNA yield from 107 WBCs was again similar for both isolation methods (62 samples in duplicate, RB yield: 6.5μg [SD 2.8] and QST yield: 7.1μg [SD 2.8]). The mean PBGD Ct value on RNA isolated with RB was 26.77, and with the QST 26.32, differing 0.55 Ct value. The PBGD quantities again demonstrated correlation between the QST and RB RNA isolation (Rcorr=0.358, data not shown), similar to the CML samples. All samples carrying a JAK2 V617F mutation, including those with low level JAK2 V617F mutation levels were clearly detectable in the QST RNA isolation (Fig. 2, patient 5 and 6).
Discussion
The isolation of high-quality RNA and DNA from various specimens is essential to perform reliable molecular diagnostic assays. In hematologic malignancies the molecular diagnostic assays are often based on RNA since the assays need to detect the chromosomal translocation-related fusion transcripts frequently present in the bone marrow or blood specimens of patients (Heim and Mitelman, 2009).
An example of such a fusion transcript is BCR-ABL1, which is present in the vast majority of patients with CML. The fusion is the result of the t(9;22) Philadelphia translocation. The favorable responses as a result of the introduction of various BCR-ABL1 TKIs and the implementation of RQ-PCR in the routine has resulted in a major increase in sampling and monitoring of CML patients based on the BCR-ABL1 fusion transcript. In fact, responses and treatment decisions are nowadays fully tuned on the BCR-ABL1 levels determined by RQ-PCR (Branford et al., 2006; Hughes and Branford 2006; Hughes et al., 2006; Cross et al., 2008). Another, acquired mutation that has recently been incorporated in the WHO classification of MPN (Vardiman et al., 2009) is the JAK2 V617F mutation (Baxter et al., 2005; James et al., 2005; Kralovics et al., 2005; Levine et al., 2005). Thus, detection of the JAK2 V617F mutation, either in RNA or DNA, is essential in the diagnosis of ET, PV, and PMF.
The increase in number of molecular analyses in MPN and CML requires automation of the routine isolation of RNA. We have used the QST for this purpose. The QIAsymphony SP instrument is especially useful for semi-high-throughput isolations of RNA and DNA. We have used a customized Qiasymphony SP RNA CT 800 V6 protocol to isolate RNA next to our routine isolations using RB. By comparing the reference gene PBGD we have shown that the PBGD levels increased after addition of BME, indicating higher quality RNA, compared with QST RNA isolated in the absence of BME. Moreover, the targets BCR-ABL1 and JAK2 V617F were detectable at similar levels to our routine analyses, only with the addition of BME. Moreover, QST RNA isolated with the addition of BME enabled BCR-ABL1 detection and JAK2 V617F detection at comparable sensitivity as RNA isolated with RB.
Of note, six samples with very low level BCR-ABL1 (<10−4.5) were detectable using RB RNA, and missed with QST RNA with BME, whereas four samples with very low BCR-ABL1 levels were detectable using QST RNA with BME, and missed with RB RNA.
Interestingly, there does not seem to be a significant difference in correlation between BCR-ABL1 levels measured in QST RNA isolated with (R=0.967) or without (R=0.995) BME after normalization with the reference gene PBGD. Apparently, this normalization corrects for the inconsistencies demonstrated by isolation of RNA with RLT Plus buffer without BME. Although the correlation at higher levels of BCR-ABL1 is similar, the sensitivity of the assay is higher in QST RNA isolated in the presence of BME as demonstrated by JAK V617F mutation detection (Fig. 2).
BME is used in some RNA isolation procedures to eliminate ribonuclease (RNAses) released during cell lysis. Numerous disulfide bonds make RNAses very stable enzymes, therefore BME is used to reduce these disulfide bonds and irreversibly denature the proteins. This prevents them from digesting the RNA during its extraction procedure (Nelson and Lehninger, 2004).
Qiagen indicates (QIAsymphony RNA Handbook, 2009) that BME may be optionally added to the buffer RLT Plus before use to ensure RNA integrity and do not recommend using BME except when isolating RNA from cells rich in RNAses. However, we clearly demonstrated that the addition of BME to this lysis buffer is essential and therefore a prerequisite in semi-high-throughput QST RNA isolation from human WBCs. Moreover, we introduced an overall modified QIAsymphony SP RNA CT 800 protocol providing micro-volumes high-quality RNA suited for reliable molecular diagnostic analyses in hematologic malignancies.
Authors' Contributions
C.P.A.M.vd.P.vd.L. performed research, analyzed data, and wrote the article. W.M.C.G.K., C.S.G., P.E.H.H., A.v.H., M.vd.W., W.T.C., and J.v.K. performed research; J.v.K. and P.J.M.V. designed research, analyzed data, and wrote the article.
Footnotes
Author Disclosure Statement
The authors reported no potential conflict of interests.