
Abstract
For inborn errors of metabolism, high resolution melting analysis (HRMA) is a rapid, efficient, simple, and inexpensive method for mutation/rare variant screening. HRMA is a recent molecular technique for genotyping single-nucleotide polymorphisms without using probes. Here we apply HRMA to the α-galactosidase a (GLA) and glucose-6-phosphatase-alpha (G6PC) genes for mutation detection of patients with Fabry disease (MIM 301500) and glycogen storage disease type 1A (GSD1A; MIM 232200), respectively. To evaluate the procedure, genomic DNAs were blindly tested for known GLA mutations (c.658C>T, c. 679C>T, c.772G>A, c.796G>A, or c.718-719delAA) in three affected males and two obligate heterozygotes with Fabry disease, a G6PC mutation (c.247C>T) in a patient homozygous for that lesion, and 10 healthy control Turkish individuals. HRMA clearly detected the mutant amplicons and discriminated them from all wild-type GLA or G6PC amplicons. HRMA proved to be a sensitive, specific, and cost-effective mutation screening method for the rapid molecular diagnosis of these inborn errors of metabolism, indicating that the technique can be readily adapted to other genetic diseases.
Introduction
S
The GLA gene, chromosomally located at Xq22.1, has seven exons and encodes the lysosomal enzyme α-galactosidase a (α-Gal A) (Chou and Mansfield, 2008; Martins et al., 2009). More than 600 GLA mutations have been reported in unrelated families with Fabry disease (Stenson et al., 2009) (www.hgmd.cf.ac.uk). The G6PC gene, localized to the chromosomal region 17q21, consists of five exons and encodes the G6PC enzyme, which catalyzes the hydrolysis of glucose-6-phosphate to glucose and phosphate in the terminal step of gluconeogenesis and glycogenolysis (Terzioglu et al., 2001; Chou and Mansfield, 2008). In this study, we evaluate the use of HRMA for the rapid molecular diagnosis of known mutations causing Fabry disease and GSD1A.
Materials and Methods
Samples and study design
Genomic DNAs from three affected males and two obligate heterozygotes with Fabry disease, and a patient with GSD1A, who were previously confirmed to have specific GLA or G6PC mutations, as well as from 10 healthy Turkish controls (five males, five females), were studied. Each sample was assigned a random digit number, and a technician blindly analyzed all samples for GLA and G6PC by HRMA. Exons that had abnormal HRMA curves were sequenced and the detected mutation was identified. Only then was the sample code broken.
DNA extraction
Genomic DNAs were extracted from peripheral blood collected in EDTA by the NucleoSpin® Blood Genomic DNA Extraction Kits (Macherey-Nagel GmbH & Co. KG, Düren, Germany) according to the manufacturer's instructions.
Gene amplification
Primer sequences for GLA and G6PC genes were previously reported (Rodríguez-Marí et al., 2003; Angaroni et al., 2004). The amplicons covered all the exons and intron-exon junctions or flanking sequences of the respective genes. PCR was performed with 15-30 ng of genomic DNA in 50 mM Tris, pH 8.3, with 10 μL MeltDoctorTM HRMA MasterMix (Applied Biosystems, Foster City, CA) containing SYTO9® Green Fluorescent Nucleic Acid Stain, and 1.2 μL of each primer at a 10 pmol/μL concentration. PCR was performed in a 7500 Fast Real-Time System (Applied Biosystems). PCR had an initial denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min.
Melting analysis
After PCR, 96-well plates were centrifuged (1500 g for 3-5 min), read in a 96-well 7500 Fast Real-Time PCR System (Applied Biosystems), and analyzed using Applied Biosystems® HRMA Software. The plates were heated at 0.1°C/s, and the fluorescence signal was recorded from 60°C to 95°C.
Sequencing
Amplicons showing abnormal HRMA profiles were sequenced using the BigDye® Terminator Cycle Sequencing Kit (Applied Biosystems) according to the manufacturer's recommendations with an ABI 3130 Genotype Analyzer.
Results
DNAs from the affected males and heterozygous females with Fabry disease, the affected patient with GSD1A, and 10 healthy controls were all blindly evaluated by HRMA. Amplicons showing abnormal HRMA profiles were then sequenced. All PCR amplifications for the GLA and G6PC genes were successful. HRMA detected abnormal melt curves for the three affected males and two heterozygotes with Fabry disease. The melt curves for the normal GLA exons were readily distinguished from those with the four GLA missense mutations and the 2 bp deletion. The normalized, derivative melt curves, difference plots, and electropherograms are shown in Figures 1-3.
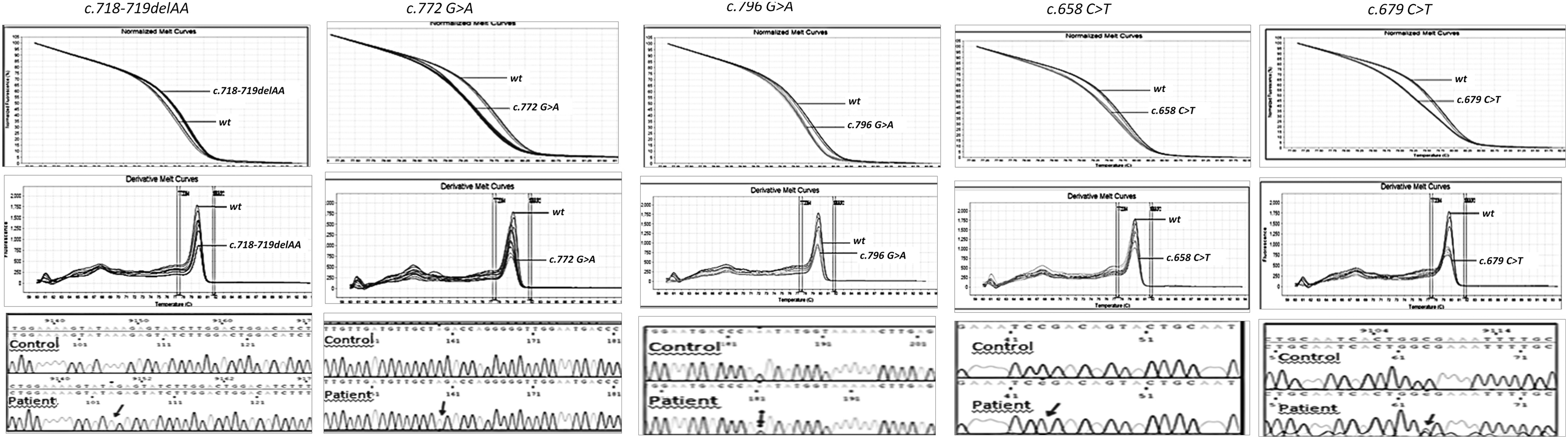
The normalized melt curves (top), derivative melt curves (middle), and electropherograms (bottom) for each α-galactosidase a (GLA) mutation. Note that mutations c. 679C>T and c.796G>A, were in heterozygous females.
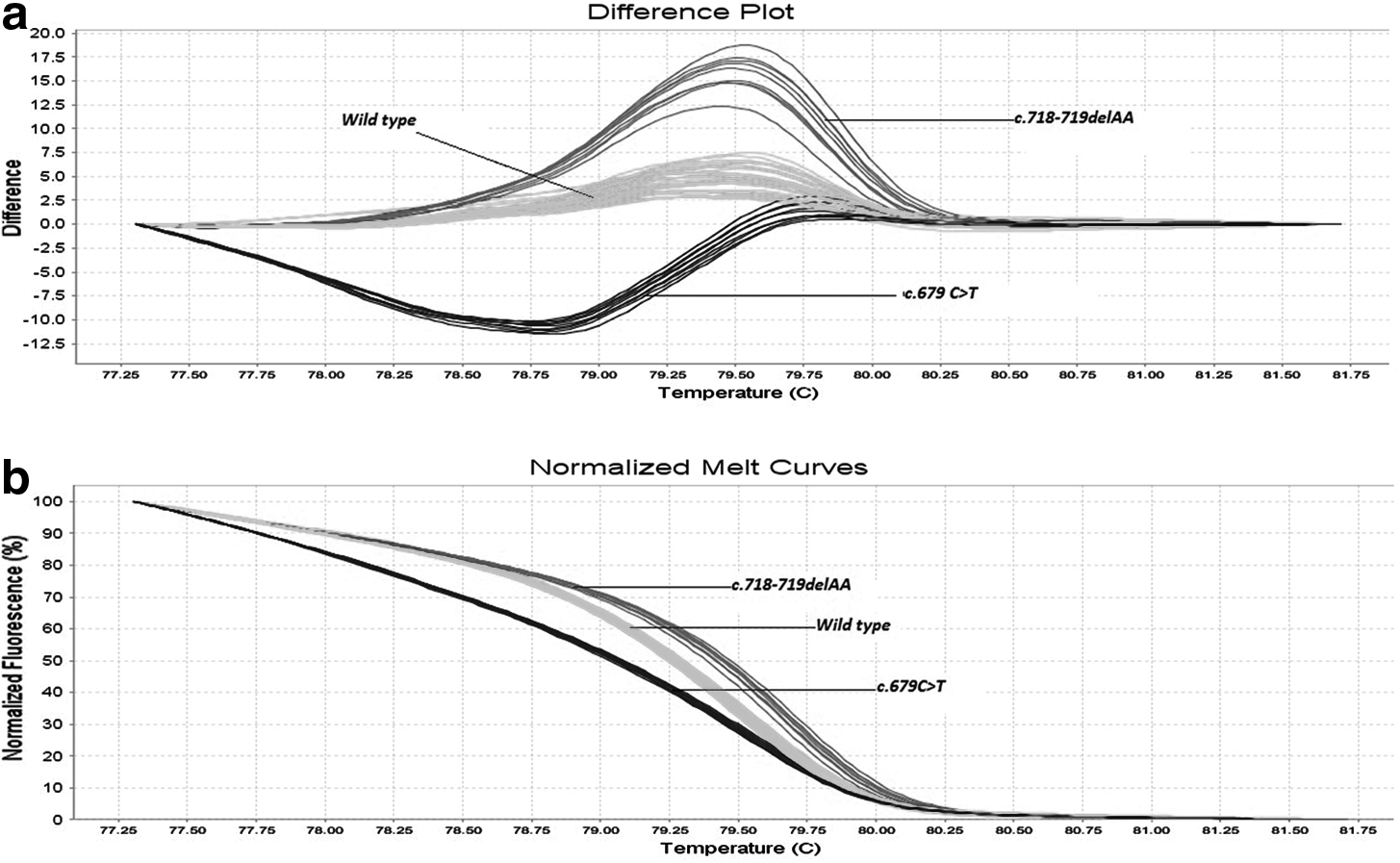

Difference plot of all the GLA amplicons carrying the mutations as well as the wild-type amplicon.
The four GLA missense mutations at different positions in the same amplicon (exon 5) were chosen purposely to investigate the sensitivity and specificity of HRMA to detect single-nucleotide polymorphisms in this X-linked gene. HRMA readily detected the two base deletion and all four exon 5 missense mutations from the wild-type amplicons. In addition, the mutations, c.679C>T and c.796G>A, in the two heterozygotes were readily detected by HRMA as shown in Figure 1.
The G6PC gene mutation, c.247C>T, in exon 2 was reported to be the most common mutation causing GSD1A among Turkish patients (Terzioglu et al., 2001). HRMA clearly identified the amplicon carrying the mutation and readily discriminated it from the GLA mutations and wild-type control amplicons. The normalized and derivative melt curves are shown in Figure 4.

Discussion
Rapid diagnosis is the most important starting point for the appropriate treatment of inborn errors of metabolism. The difficulty in establishing and performing the assays, the duration and the need for having the right sample from the right tissue, even sometimes requiring biopsies, are the disadvantages of biochemical methods (Longo, 2006; Ezgu et al., 2008). Although the molecular methods, especially DNA sequence analysis, provide accurate diagnosis and do not need tissue samples other than whole blood from which DNA can be extracted easily, the cost of the analysis has made researchers look for rapid molecular screening techniques. Previously, several methods such as DHPLC, DGGE, and SSCP for presequencing screening have been used effectively to identify amplicons for resequencing (e.g., Sevilla et al., 2002; Tabone et al., 2006). Recently, HRMA was employed successfully for mutation screening (Vossen et al., 2009).
In this study, we evaluate HRMA to rapidly and inexpensively screen for mutations in X-linked Fabry disease and autosomal recessive GSD1A. HRMA of amplicons depends on their DNA melting curves in the presence of saturating DNA binding dyes. As the temperature of the solution is increased, the specific sequences of the amplicon (primarily the GC content and the length) determine the melting behavior. When the fluorescence signal is plotted against the temperature, the fluorescence intensity decreases as the double-stranded DNA becomes single stranded and the dye is released. The melting temperature (Tm) at which 50% of the DNA double stranded is calculated by taking the derivative of the melting curve (Erali et al., 2008; Vossen et al., 2009).
In this study, the application of HRMA to two inborn errors of metabolism, Fabry disease and GSD1A, was investigated, as their respective biochemical diagnoses have significant challenges. For Fabry disease, the determination of leukocyte α-Gal A activity in males is diagnostic, but heterozygous females can have very low to high normal activities due to random X-inactivation (Rodríguez-Marí et al., 2003). Thus, sequencing the GLA gene and the identification of disease-causing mutations is required for the accurate diagnosis of suspect female heterozygotes and for genotype/phenotype correlations in affected males. Given the fact that enzyme replacement therapy is available, molecular diagnosis is essential for confirming all affected males and to accurately identify heterozygotes (Zarate and Hopkin, 2008; Germain and Fan, 2009). Previously, presequencing mutation screening of the GLA gene with DHPLC was reported (Shabbeer et al., 2005). HRMA of the GLA gene clearly proved effective in identifying the amplicons that carry the mutations, even in ones that lie very close to each other in both affected males and heterozygous females. HRMA discrimination was not only clear in affected males, but also in the amplicons from female heterozygotes. In addition, the 2 bp deletion in exon 5 was clearly demonstrated.
The biochemical diagnosis of GSD1A relies on the determination of glucose-6-phosphatase levels from liver samples obtained by biopsy. The need for hospitalization to perform this invasive method as well as the need for rapid diagnosis for this treatable disorder also makes molecular diagnosis preferable over biochemical methods. Previously, DHPLC for presequencing screening and microarray technology for screening for common mutations have been used for G6PC mutation analyses (Forsyth et al., 2005; Xu et al., 2010). In this study, HRMA of the G6PC gene was performed on genomic DNA from a previously confirmed GSD1A patient. Although others have noted difficulty in detecting homozygous mutants from the wild-type profile even with HRMA (Erali et al., 2008), homozygosity for the c.326C>T mutation was clearly discriminated from the normal control samples.
Compared with other presequencing mutation screening methods such as DHPLC, HRMA may be more sensitive and specific, even for homozygous mutant samples. HRMA is a closed-tube method, which can be directly performed after PCR without any additional procedural steps. In addition, it is a much more rapid technique compared with other mutation screening methods. Another advantage of HRMA is that after completing the analysis, the same samples can be directly used for sequencing, thereby eliminating the need for a new amplification reaction (Chou et al., 2005; Aguirre-Lamban et al., 2010). Finally, the lower cost for mutation detection emphasizes the advantages of HRMA for mutation screening. Thus, HRMA should be considered when screening in other genetic diseases.
Footnotes
Acknowledgments
These studies were supported, in part, by the Department of Genetics and Genomic Sciences of the Mount Sinai School of Medicine, NY.
Author Disclosure Statement
The Mount Sinai School of Medicine, the Department of Genetics and Genomic Sciences and some faculty members in the department (including the Dean for Genetic and Genomic Medicine, Dr. Robert Desnick, the Principal Investigator in this study), receive financial benefit from the Genzyme Corporation for the sale of Fabrazyme®, an enzyme replacement drug developed by them for the treatment of Fabry disease.