
Abstract
Deafness encompasses a series of etiologically heterogeneous disorders with mutations in more than 400 independent genes. However, several studies indicate that a large proportion of both syndromic and nonsyndromic forms of deafness in the racially diverse Indian and Pakistani populations are caused by defects in just a few genes. In these countries, there is a strong cultural preference for consanguineous marriage and an associated relatively high prevalence of genetic disorders. The current Indian population is approximately 1.2 billion and it is estimated that 30,000 infants are born with congenital sensorineural hearing loss (HL) each year. The estimated rate of profound bilateral HL is 1.6 per 1000 in Pakistan and 70% of this HL arises in consanguineous families. Knowledge of the genetic cause of deafness within a distinct population is important for accurate genetic counseling and early diagnosis for timely intervention and treatment options. Many sources and technologies are now available for the testing of hearing efficiency. Population-based screening has been proposed as one of the major strategies for translating genetic and genomic advances into population health gains. This review of the genetics of deafness in Indian and Pakistani populations deals with the major causes of deafness in these countries and prospectives for reducing the incidence of inherited deafness.
Introduction
H
Despite the obstacles of locus and allelic heterogeneity, genetic screening to identify genes and specific variants that contribute to NSHL has been widely implemented worldwide, especially with regard to identifying the spectrum of genes and variants associated with deafness across various populations.
The genetic structure of human populations is mainly determined by geography, history, and culture. The marriage patterns have significant implication for these factors underlying the biological phenomenon over time (Burchard et al., 2003). In particular, interfamily marriages are recognized as being associated with a higher risk of passing an autosomal recessive genetic disorder (Al-Gazali, 2005; Bittles, 2005a). It is indeed estimated that about 10% of congenital and genetic disorders worldwide are associated with customary consanguineous marriage (WHO, 2000). In addition, populations that have a small number of founders and/or are geographically or culturally isolated from other populations have been a precious resource for genetic mapping studies of inherited disorders (Arcos-Burgos and Muenke, 2002; Heutink and Oostra, 2002).
The huge population of the Indian subcontinent (Pakistan, India, and Bangladesh) has provided an opportunity for studies of genetic disorders (Gadgil et al., 1998). Due to their unique geography and history, the population of India (over 1.21 billion) and Pakistan (approximately 140 million) is a goldmine for these studies. In addition, it is an admixture of ethnically diverse populations with unique familial and social structure (Mehdi et al., 1999). From a genetic perspective, the population of India is unique, because of the traditional status consanguineous unions regarded as customary for the people of Southern India. For example, in Tamil Nadu, a South Indian state, 47% of all marriages were consanguineous, compared to 25% in Maharashtra (West India) and less than 10% of marriages in the Northern and Eastern states of India were consanguineous (IIPS, 1995). It is estimated that as many as 45-60% of marriages in Pakistan are between close relatives (the vast majority of marriages are between first cousins) (Bittles, 2005b).
Consanguineous families have contributed significantly to the identification of mutated genes associate with HL (Friedman and Griffith, 2003). Autosomal recessive forms (DFNB) of isolated deafness are higher than the world average in the Indian and Pakistani populations due to a preponderance of consanguineous marriage, caste barriers in marital relations, and assortative mating, whereas autosomal dominant, X-linked, and mitochondrial inheritance of deafness are less prevalent (Bhalla et al., 2009; Padma et al., 2012). In this study, we review the genetics of familial deafness in Indian and Pakistani populations. A summary of these genes is given in Tables 1 and 2, and more about the most common mutations causing HL is discussed in the text. Genes and geographic maps of population sampling locations for both countries are shown in Figures 1 and 2.
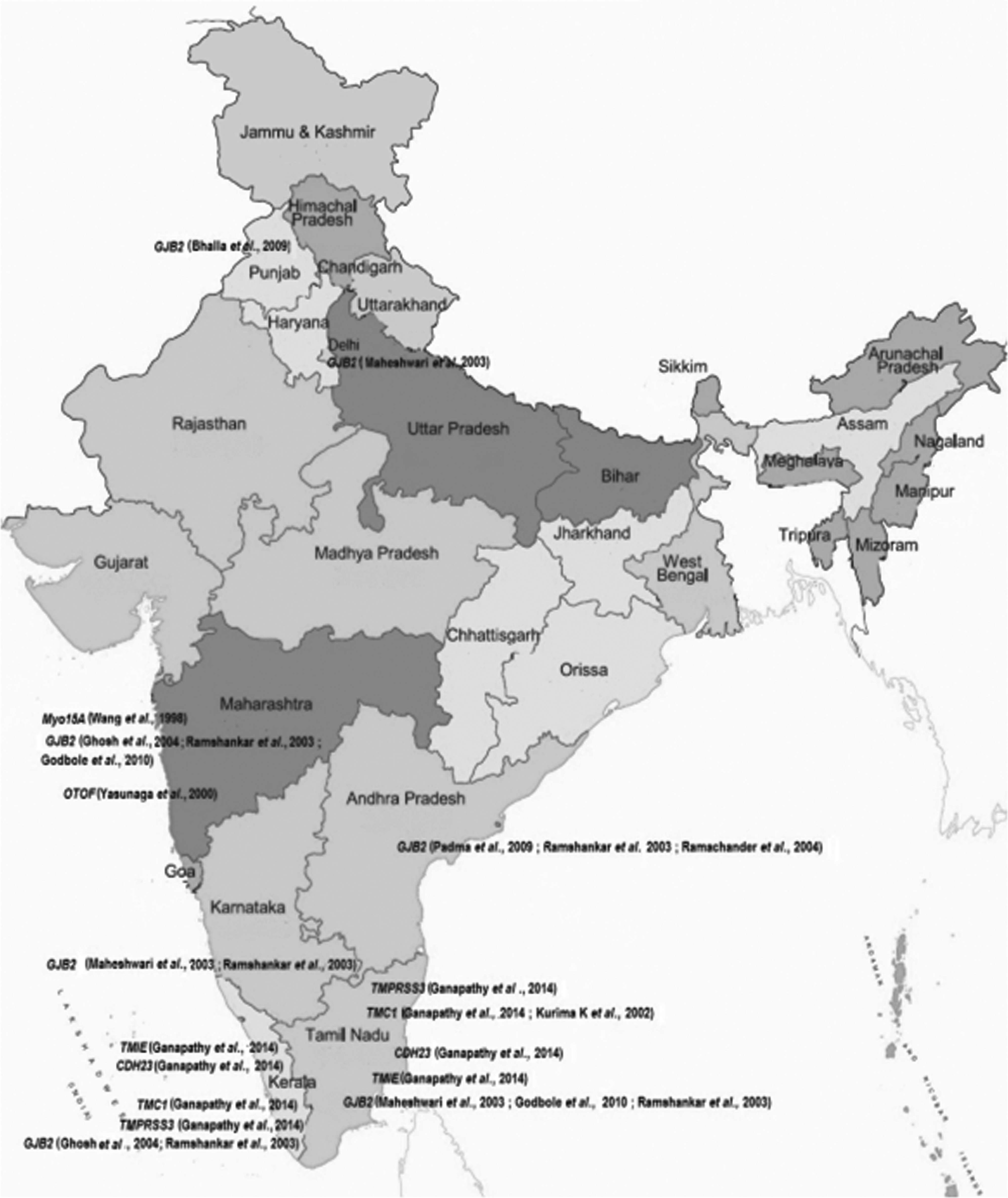
Genes and geographic maps of Indian population sampling locations.
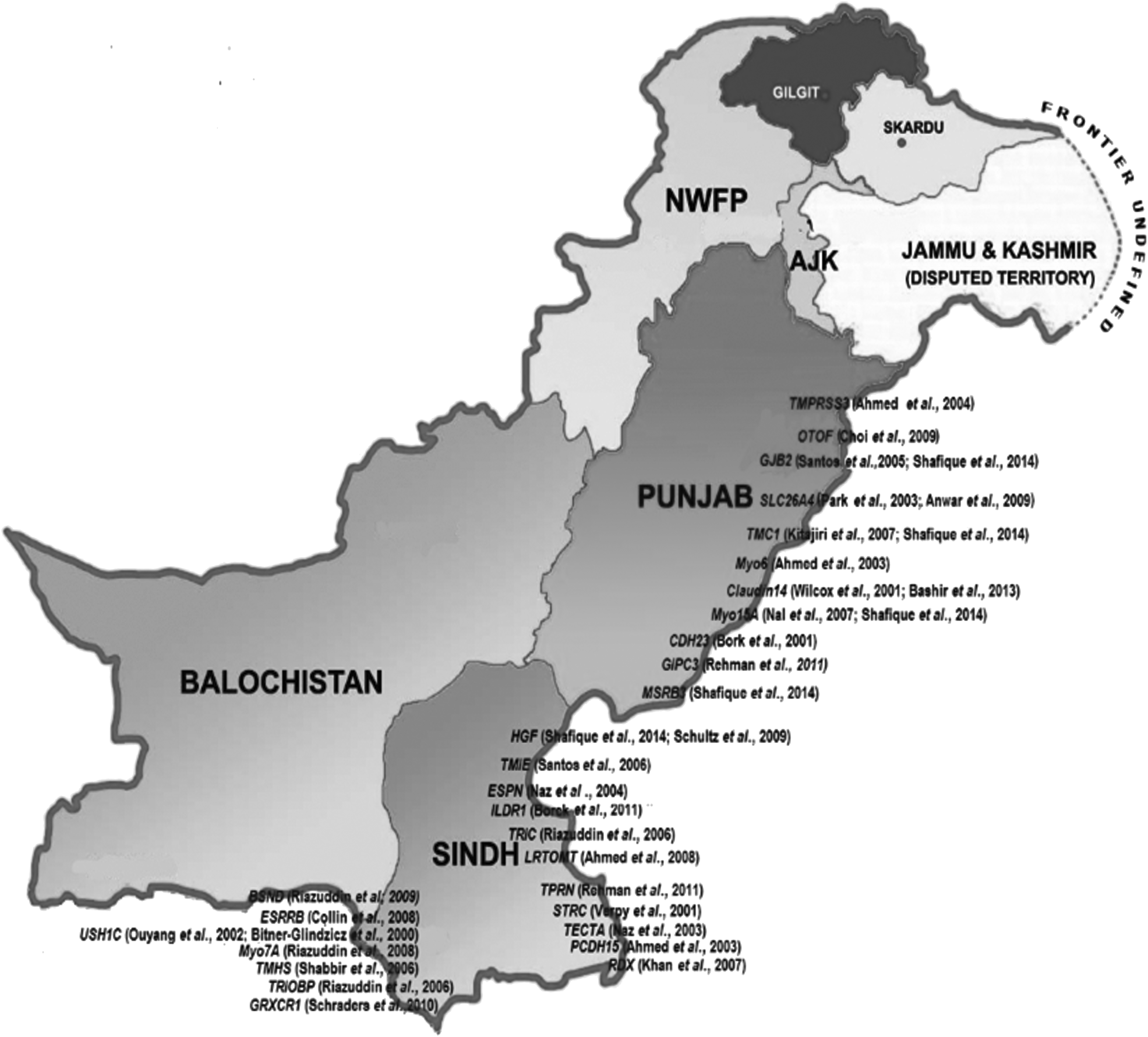
Genes and areas of samples collection in Pakistan. NWFP, North West Frontier Province.
Bhalla et al. (2009); 2Padma et al. (2009); 3Godbole et al. (2010); 4 Ramshankar et al. (2003); 5 Mani et al. (2009); 6Joseph and Rasool (2009); 7Wang et al. (1998); 8Ali et al. (2013); 9Yasunaga et al. (2000); 10Ganapathy et al. (2014); 11Kurima et al. (2002); 12Ahmed et al. (2002); 13Naz et al. (2002); 14Rizuddin et al. (2006); 15Shultz et al. (2009); 16Charizopoulou et al. (2002).
Amino acids.
Ahmed et al. (2004); 2Choi et al. (2009); 3Santos et al. (2005); 4Shafique et al. (2014); 5Park et al. (2003); 6Anwar et al. (2009); 7Kitajiri et al. (2007); 8Ahmed et al. (2003a); 9Wilcox et al. (2001); 10Bashir et al. (2013); 11Nal et al. (2007); 12Bork et al. (2001); 13Rehman et al. (2011); 14Schultz et al. (2009); 15Santos et al. (2006); 16Naz et al. (2004); 17Borck et al. (2011); 18Riazuddin et al. (2006a); 19Ahmed et al. (2008); 20Rehman et al. (2011); 21Verpy et al. (2001); 22Naz et al. (2003); 23Ahmed et al. (2003b); 24Khan et al. (2007); 25Schraders et al. (2010); 26Riazuddin et al. (2006b); 27Shabbir et al. (2006); 28Riazuddin et al. (2008); 29Ouyang et al. (2002); 30Bitner-Glindzicz et al. (2000); 31Collin et al. (2008); 32Riazuddin et al. (2009).
Amino acid.
Most of the DFNB forms cause a phenotypically identical severe to profound, prelingual nonprogressive HL. However, mutations at a few loci, DFNB1 (GJB2), DFNB3 (MYO15A), and DFNB29 (Claudin 14), can result in variation in severity even within families (Cohn et al., 1999; Bashir et al., 2012, 2013). This is suggestive of other factors modifying the effects of mutations in the involved genes. Deafness is usually stable, but progression has been reported. DFNB2 (MYO7A) and DFNB8 (TMPRSS3) cause a delayed childhood-onset HL. Also of note is that HL in DFNB4 due to mutations in SLC26A4 may be associated with dilated vestibular aqueducts and endolymphatic sacs, and there is a link of auditory neuropathy with mutations in DFNB9 (OTOF). In addition, vestibular symptoms have been noted in DFNB2 (MYO7A), DFNB4 (SLC26A4), and DFNB12 (CDH23) (Bitner-Glindzicz, 2002).
The overall incidence of GJB2 mutations in the Indian population is around 25%. Several other DFNB genes to recessive, severe to profound, congenital, or prelingual-onset deafness in the Indian population include TMC1 (DFNB7/11 (1.6%), CDH23/DFNB12 (1.8%), DFNB8/10/TMPRSS3 (1.2%), and TMIE (1.6%) (Table 1). In the Pakistani population, 6.1-53% of inherited deafness is caused by mutations in GJB2. Other important contributors to genetic deafness are SLC26A4 (7.2%), OTOF (2.3%), MYO15A (3.3-13%), TMC1 (3.4-5.4%), TMPRSS3 (1.8-2.5%), and TMIE (1.7%) (Table 2).
Nonsyndromic Recessive Forms of Deafness in Indian Families
The prevalence of deafness in India is fairly significant. It is the second most common cause of disability. Approximately 63 million people (6.3%) in India suffer from significant auditory loss, based on population surveys in 2003 using the WHO protocol (WHO, 2009a). The estimated prevalence of adult-onset deafness was found to be 7.6% and childhood-onset deafness was found to be 2%. Molecular genetic epidemiology studies have revealed that deafness is widely prevalent in the country, especially in Southern India where the incidence is six times that of the global average.
DFNB1/GJB2/GJB6
Gap-junction proteins expressed in the cochlea have been shown to be essential for normal hearing as they are involved in the recycling of potassium ions that flow into sensory hair cells during the process of mechanotransduction. Mutations in the GJB2 (gap-junction protein beta 2) gene that encodes the connexin-26 protein have been unambiguously cited as the leading cause of nonsyndromic prelingual congenital deafness throughout the world, with certain mutations having higher incidences in specific populations. For instance, the 35delG mutation is the leading genetic cause of deafness in Europe and the United States (Lucotte and Mercier, 2001; Roux et al., 2004; Lucotte and Dieterlen, 2005; Putcha et al., 2007). Instead, the 235delC mutation has been observed widely in patients from East Asia (i.e., China, Japan, and Korea) (Abe et al., 2000; Ohtsuka et al., 2003; Yan et al., 2003). Other deafness-associated GJB2 mutations include 167delT in Ashkenazi Jews and the R143W in Africans (Morell et al., 1998).
The overall incidence of GJB2 mutations in cohorts of Indian family studies was observed to be 22.5%, with a regional variation from 15.4% in Western (Maharashtra) to up to 36% in Southern (Kerala) parts of India. Biallelic W24X was being the major mutation identified. Its carrier frequency was estimated to be 0.024 (RamShankar et al., 2003; Godbole et al., 2010; Nayyar et al., 2011). A possible founder effect for this mutation in the Indian population has subsequently been suggested (RamShankar et al., 2003). The W24X mutation typically manifests in the majority of deaf patients across most regions of India. However, North India seemed to have a lower prevalence of biallelic W24X mutations. In addition, W77X and Q124X appeared to be more common in the north (Ghosh et al., 2004). The variation in the incidence of the W24X mutation between the northern and southern populations is further highlighted by a study by Ramchander et al. (2004).
The 35delG mutation has also been known to occur in the heterogeneous Indian population (Mani et al., 2009). In a cohort of North Indian patients, the 35delG mutation has indeed been found to surpass the occurrence of W24X, accounting for close to 10.9% incidence over the erstwhile predominant W24X (3.8%), confirming the genetic complexity that is the result of extensive genetic diversity in the Indian population (Bhalla et al., 2009).
Ethnic differences between the South (indigenous Dravidians) and North Indians (Indo-European migrants from Russia) based on linguistic genealogy have also been discussed (Passarino et al., 1996). This theory has been reinforced by studies on South Indian patients showing variation in the occurrence of W24X ranging from 32.5% to 72.72% in deaf patients with GJB2 mutations (Joseph and Rasool, 2009; Padma et al., 2009). W24X has also been found in 4-5% of 603 unrelated indigenous Romanies believed to have once migrated to the Balkan and Vlax Romani territories from India (Bouwer et al., 2007). Reports from Slovakia (Minarik et al., 2003) and Spain (Alvarez et al., 2005) indicate 39% occurrence of W24X, consolidating the theory of emigration of the ancestral Romanies from India and a consequent founder effect.
W77X is the second most frequent mutation with affected individuals dispersed in most regions in India. Incidences for this mutation vary from 0.5% (Padma et al., 2009) to 5.9-6.89% (Bhalla et al., 2009; Godbole et al., 2010) of patients with GJB2 mutations. Q124X has also been detected in the Indian population and with W77X appeared to be more common in North India. The 235delC mutation detected in 0.5-3% of deaf patients also has been documented in Southern India (Ramchander et al., 2004; Padma et al., 2009). Occurrence of the IVS1+1G>A mutation among Indians at a prevalence of 0.3-4.68% was documented in a study from Southern India (RamShankar et al., 2003) (Table 1).
An in-frame deletion, delE120 in GJB2, was first identified in a consanguineous Indian Muslim family and has been shown to occur in 0.8-1.7% of patients, pointing to a higher prevalence in the North (Bhalla et al., 2009; Mani et al., 2009). The mutation was subsequently reported in Pakistan, Germany, and Turkey. Other potentially pathogenic mutations found include the R143W mutation, I35S, I33T, and W172R (Mani et al., 2009).
Surprisingly, an absence of GJB6 mutations has been reported in North Indian patients, in a study aiming to find individuals with DFNB1, due to GJB6 mutations concordant with the high allele frequency of 35delG observed previously in a similar cohort (Bhalla et al., 2009).
DFNB3/MYO15A
Mutations in the gene coding for myosin XVA cause DFNB3 (Wang et al., 1998; Belyantseva et al., 2005). DFNB3/MYO15A is not a common cause of deafness in the Indian population with 600 families screened, only 6 showed linkage to DFNB3, thus contributing only 1% (Ali et al., 2013). Studies in mice suggest that MYO15A is necessary for actin organization in the hair cells of the cochlea and its interactions with other proteins, besides WHIRLIN, are important for hair cell maturation (Mustapha et al., 2007). Originally mapped in deaf individuals born in the village of Bengkala in Bali (Wang et al., 1998), DFNB3 has subsequently been shown to segregate in two unrelated deaf Indian families. The haplotypes of affected individuals in these two Indian families were different from each other and different from the Bengkala haplotype, suggesting that these three DFNB3 mutations arose independently. The two mutations identified in the Indian families include N890Y and K1300X (Table 1).
DFNB8/DFNB10/TMPRSS3
The gene encoding a transmembrane serine protease, TMPRSS3, has been shown to be responsible for both the DFNB8 and DFNB10 phenotypes (Scott et al., 2001). The deafness DFNB8 locus was mapped by linkage analysis on a large Pakistani family consisting of 41 members (Veske et al., 1996). Studies by Guipponi et al. (2002) suggested the existence of autocatalytic processing, by which TMPRSS3 would become active, and the epithelial sodium channel (ENaC) could be a substrate of TMPRSS3 in the inner ear. Its contribution to hereditary deafness in the Indian population has recently been evaluated in a screening of 1739 individuals from 374 families with NSHL (Ganapathy et al., 2014). Analysis led to the identification of 3 TMPRSS3 novel mutations, including p.V116M, p.G243R, and p.C386R. TMPRSS3 mutations have been noted as the cause of deafness in 1.2% of the families included in this study.
DFNB7/11/TMC1
Mutations in the transmembrane channel-like 1 (TMC1) gene cause DFNB7/11. All TMC family members contain a conserved 120-amino acid sequence, which is called the TMC domain. TMC proteins may act as ion channels, ion pumps, or transporters depending on their structure. Data obtained from the tmc1 mutant mice suggest a possible role for TMC1 in mechanoelectrical transduction of sound by cochlear hair cells (Kawashima et al., 2015). In their screening of 374 families, Ganapathy et al. (2014) identified eight TMC1 mutations. Among these, five were novel. The identified variants include the splice site mutations c.237-6T>G, c.453+2T>C, c.1566+1G>A, the deletion mutation p.I210del, and the missense mutations (p.V372M, p.R445C, and p.G267E). The nonsense mutation p.R34X, a commonly occurring mutation in multiple world populations (saïd et al., 2010), was also observed in one family. TMC1 mutations have been pinpointed as the cause of deafness in 1.6% of the families included in this study.
DFNB6/TMIE
Mutations in the transmembrane inner ear-expressed gene (TMIE) cause DFNB6. Studies in mice suggest that this gene is required for normal postnatal maturation of sensory hair cells in the cochlea, including correct development of stereocilia bundles, essential for mechanotransduction of sound (Chung et al., 2007). Ganapathy et al. (2014) identified three known TMIE mutations in their study. These include two missense mutations (p.E31G and p.R84W) and one insertion mutation (c.125_126insCGCC). In this cohort, HL is attributed to these TMIE mutations in 1.6% of families.
Nonsyndromic Recessive Forms of Deafness in Pakistani Families
The Pakistani population is one of the richest genetic sources to study hereditary disorders due to a mixture of diverse ethnicities with unique familial and social characteristics (Mehdi et al., 1999) and its unique sociocultural trends, where out of every ten marriages, six are consanguineous and four are between first cousins (Hussain and Bittles, 1998). It is estimated that the prevalence of hearing impairment in Pakistan is 1.6 per 1000 live births (Elahi et al., 1998), which is higher than the world average of 1 per 1000 live births (Atar and Avraham, 2005).
DFNB1/GJB2
The contribution of GJB2 variants to NSHL in the Pakistani population has been reported to vary from 6.1% to 50%. Screening of 430 affected subjects from 196 families for GJB2 variations by Santos et al. (2005) showed that only 6.1% of the families had GJB2-related HL. W24X and W77X were found to be the most common. The 167delT, R2H, delE120, and L90P mutations have also been detected. Of the families that were studied, 70% come from the Punjab province, the largest province in Pakistan. The native languages of these families are Punjabi and Saraiki. The remaining 30% of the families in the study were equally representative of the other provinces and their corresponding language groups. There was no association found between the presence of a particular GJB2 mutation and the region of origin or linguistic background. A recent study by Bukhari et al. (2013) also established that GJB2 does not contribute to a high proportion of isolated HL in the Hazara population. Hazara is a province of Khyber Pakhtunkhwa of Northern Pakistan. Their sequencing analysis of 70 patients led to the identification of three GJB2 mutations in three individuals (4.28%). These include p.W24X and two other mutations (p.I35S and c.35delG) identified for the first time in Pakistan. However, recent studies indicate that the frequency of GJB2 mutations is higher in Pakistani deaf patients; of the 20 large consanguineous families from different areas of Pakistan screened for GJB2, 10 (50%) were reported to have GJB2-related HL (Anjum et al., 2014). A total of six different mutations were identified in the study, with W24X as the major mutation found, followed surprisingly by c.35delG being the second most prevalent, surpassing Q124X, W77X, V153I, and T8M. Furthermore, sequence analysis of GJB2 in 30 consanguineous families with NSHL from different regions of Punjab, by Shafique et al. (2014), revealed the presence of mutations in this gene in 16 families (53%). W24X and W77X were the most common mutations found. In addition, a novel homozygous change p.G200R was detected in one family. Other known mutations identified include c.35delG, E147K, and c.377_378insATGCGGA (R127Cfs*85).
DFNB9/OTOF
Mutations in OTOF (DFNB9), encoding otoferlin, usually lead to prelingual profound deafness (Yasunaga et al., 2000). OTOF-related HL can also be associated with intact otoacoustic emissions as part of a distinctive phenotype referred to as auditory neuropathy/dyssynchrony (AN/AD) (Varga et al., 2003). Otoferlin has been proposed to act as a calcium sensor that regulates synaptic vesicle exocytosis in cochlear hair cells (Roux et al., 2006). Most of the pathogenic alleles of OTOF are private, each one being reported in only one family. A total of 13 (2.3%) families linked to DFNB9 were identified in a screening of 557 large consanguineous Pakistani families (Choi et al., 2009). Probable pathogenic variants in the OTOF gene were found among all 13 families. These include the previously reported nonsense mutation p.R708X (Rodriguez-Ballesteros et al., 2003) and 10 unreported variants: 3 nonsense mutations (p.R425X, p.W536X, and p.Y1603X), 1 frameshift (c.1103_1104delinsC), 1 deletion mutation (p.E766del), and 5 missense substitutions (p.L573R, p.A1090E, p.E1733K, p.R1856Q, and p.R1939W). A novel splice site variant (c.766-3C>T) in the cis configuration with p.E766del was also found in one Pakistani family (Table 2).
DFNB4/SLC26A4
DFNB4/PDS is the most common locus in the Pakistani population with a prevalence of 7.2% (Anwar et al., 2009). Pendrin is the transmembrane protein encoded by SLC26A4 that functions primarily as a transporter of chloride and iodide across the membranes of the cell. In the cochlea, SLC26A4 is mainly expressed in regions of endolymph resorption; therefore, mutations in SLC26A4 may disturb endolymphatic fluid homeostasis (Borck et al., 2003). Anwar et al. (2009) analyzed the SLC26A4 gene in 46 large consanguineous Pakistani families linked to DFNB4/PDS. Their study revealed 16 probable pathogenic variants, 8 of which were not previously reported. These include three missense substitutions (p.R24L, p.G139V, and p.V231M), two splice site variants (c.304+2T>C and c.1341+3A>C), and one frameshift (c.1692_1693insA) resulting in premature protein (p.C565MfsX8) truncation. Each of the six pathogenic variants (p.V239D, p.Q446R, p.S90L, p.Y556C, p.R24L, and p.K715N) was found in more than one family and haplotype analyses suggest that they are founder mutations. Two genomic deletions were also identified: the c.-23177_c.164+1027del24368ins7 and c.1264-477_2090-4927del11202. Eight earlier reported pathogenic variants (p.V239D, p.S90L, p.I455F, p.Y556C, p.Q446R, p.A372V, p.S57X, and p.K715N) were also observed (Reardon et al., 1997; Park et al., 2003; Tsukamoto et al., 2003) (Table 2).
DFNB7/11/TMC1
Mutations in the TMC1 gene cause DFNB7 (Fukushima et al., 1995; Kurima et al., 2002). Kitajiri et al. (2007) reported 9 different mutations in the TMC1 gene in 19 (3.4%) of the 557 Pakistani families with NSHL (Table 2). A single mutation p.R34X causes deafness in 10 (10/557; 1.8%) of the families. A shared haplotype in different families strongly suggests a single ancestral origin of the mutation. Other variants were also detected, including IVS5+1G>T, p.P514L, and p.C515R. Santos et al. (2005) reported a prevalence of 4.4% of NSHL due to TMC1 in the Pakistani population. We previously estimated that TMC1 mutations account for approximately 5% of recessive deafness in a mixed cohort of 230 Pakistani and Indian families (6/10 Punjabi families) (Kurima et al., 2002). Of the 30 families analyzed by Shafique et al. (2014), 3 (10%) were found to carry TMC1 mutations.
DFNB29/CLDN14
Deafness at the DFNB29 locus is caused by recessive mutations of CLDN14, encoding claudin 14, a tight junction protein. Loss of claudins has been reported to result in increased paracellular permeability of K+ in the reticular lamina and an elevation in the K+ concentration around the basolateral regions of hair cells, which is toxic. Thus, claudins likely act as selective electrostatic barriers for anions and cations. The relative contribution of CLDN14 mutations in the Pakistani deaf population is approximately 2.25% (18/800 families) and is associated with marked inter- and intrafamilial variability in hearing thresholds (Bashir et al., 2013). The identified mutations include two unreported missense substitutions (p.S87I and p.A94Val) and three known variants (p.R81H, p.V85D, and p.M133RfsX23) (Wilcox et al., 2001). Haplotype analyses indicate that p.V85D and p.M133RgfsX23 are founder mutations. The p.V85D accounts for approximately 67% of the mutant alleles of CLDN14 in the study.
DFNB3/MYO15A
DFNB3 is responsible for 3.3-13% of recessive deafness in the Pakistani population (Nal et al., 2007). A total of 17 mutations (IVS4+1G>T, Q2716H, Q1229X, p.T1253fsX1277, p.Y1392X, p.K1557E, p.G1706V, p.L1730P, p.G2018R, p.Q2021X, p. T22051, p.G2244E, p.V2266M, p.2720H, p.V2940fsX3034, p.L3160F, and p.Q3492X) were identified in Pakistani deaf patients in the MYO15A gene by two independent groups (Liburd et al., 2001; Nal et al., 2007). These seventeen missense, nonsense, and splice site mutations are located in exons encoding motor and first FERM domains of MYO15A. In a more recent study, 4/30 (13%) families were found to have mutation in MYO15A (Shafique et al., 2014). These include four unreported variants, of which two were splice site mutations (c.9948G>A and c.3866+1G>A), one was a nonsense (p.R2923X), and one missense mutation (p. F2741S). Interestingly, Bashir et al. (2012) reported a duplication of cytosine in a stretch of seven repetitive C nucleotides (c.1185dupC) in exon 2 of MYO15A, leading to a frameshift mutation (p.E396fsX431), which was identified in a Pakistani family with moderate to severe HL. A less severe HL with some residual hearing at low frequencies or moderate deafness has only been reported if mutations in MYO15A lie in the second exon of this gene (Nal et al., 2007; Cengiz et al., 2010). A role of modifier genes in determining the HL phenotype has also been proposed since individuals with identical mutations in exon 2 of MYO15A may exhibit different degrees of HL (Liburd et al., 2001; Nal et al., 2007).
Conclusion
Work during the past decade has generated a wealth of information connecting mutations with health. This has dramatically enhanced our understanding of the pathogenetic basis of inherited diseases. However, due to extreme clinical and genetic heterogeneity, full comprehension of these correlations still remains a great challenge in the case of NSHL. Studies have been particularly successful when investigating the fundamental genetic cause of NSHL in families from regions of the world with high consanguineous marriage rates. Despite progress made in the identification of deafness genes, the population distribution for many of the newly identified HL genes still remains unclear. It is well known that the molecular epidemiology of deafness can vary considerably among populations.
We have summarized the various genes and mutations associated with HL in the Indian and Pakistani populations. In these studies, the spectra of sequence variations in the deafness genes were frequently different from those observed outside the Indian subcontinent. For instance, the W77X variant in GJB2 has only been reported in people originating from the Indian subcontinent. The W24X variant has been observed in Europeans, but its prevalence is about three times higher in Pakistanis and at least 20 times higher in Indians. Thus, the occurrence of the same deafness loci and spectrum of variants in several genes in both populations may reflect shared origins of HL alleles within the Indian subcontinent. The reports also show that there is a need for awareness and accurate genetic counseling to families with high genetic risk, for which it is necessary to establish genetic testing for the common deafness mutations prevalent in the ethnic populations. A promising technique for genetic testing is the use of DNA microarrays. This approach offers the possibility of testing for many known mutations and/or a complete mutation analysis of one or more genes in parallel in a single experiment.
Homozygosity mapping has proven successful with consanguineous families and homogeneous populations sharing similar clinical features. However, due to limiting meiotic crossing-over events, these locus mapping approaches typically identify large chromosomal regions that include hundreds of genes. These loci could now undergo parallel sequencing of all linked genes through either custom targeted capture and massively parallel sequencing of the single locus or whole-exome sequencing (WES). In addition, small families unsuitable for conventional approaches can now be alleviated through the use of next-generation sequencing powered methods such as WES.
Deafness remains a high burden for the Indian subcontinent. A lack of skilled manpower and human resources makes this problem a huge challenge. According to the WHO (2009b) statistics, half the causes of deafness are preventable in India. About 30%, although not preventable, are treatable or can be managed with assistive devices. To address the issue of deafness, the Government of India launched the National Programme for Prevention and Control of Deafness in 2006, with a focus on manpower development for prevention, early identification, and ear service provision, including rehabilitation (Garg et al., 2009). Since the programme is also being implemented at the primary healthcare level, it envisages a reduction in the burden of deafness and prevention of future HL in India.
Footnotes
Acknowledgment
This work is supported by the National Institutes of Health (NIH) (grant R01DC005575, R01DC012546, and R01DC012115) to X.Z.L.
Author Disclosure Statement
The authors declare no conflicts of interest.