
Abstract
Background: Myotonic dystrophy (DM) is the most common adult form of muscular dystrophy, characterized by autosomal dominant progressive myopathy, myotonia, and multiorgan involvement. Myotonic dystrophy type 2 (DM2) is caused by a [CCTG] expansion in the ZNF9/CNBP gene. The aim of this work was the validation of the new molecular diagnostic test Myotonic Dystrophy type 2 kit-FL. Results: A cohort of 126 individuals was analyzed. The results show that 126/126 patients were correctly identified using the new molecular assay. In particular, 74 were DM2 positive, 39 were DM2/DM1 negative and 13 DM2 negative/DM1 positive. Approximately 9.5% (7/74) of the DM2-positive samples had a single sizeable expansion and 85% (63/74) showed multiple bands or smears. Comparative fluorescence in situ hybridization (FISH) analyses, on muscle biopsies, revealed that the sensitivity and specificity were very high (>99%). Equivalent analytical performances were obtained using different DNA extraction methods. Among affected individuals 87.5% (28/32) had electrical myotonia, 69% (22/32) proximal weakness, 41% (13/32) cataracts, and about 37.5% (12/32) cardiac conduction defects. FISH analysis and clinical data were used to support the genetic analysis.
Introduction
M
In the European population, prevalence of DM2, is not well established, but estimated to be similar to DM1 (Moxley et al., 2002; Udd et al., 2006). Although DM1 and DM2 have similar symptoms, they also present a number of very dissimilar features making them clearly separate diseases (Day et al., 2003; Cho and Tapscott, 2007; Meola, 2013). The most important differences are absence of a congenital form and the lack of anticipation in DM2 disease (Udd et al., 2003). Indeed in DM2 the mutation usually contracts in the next generation. The range of the [CCTG] repeat is below 30 repeats in normal individuals while the range of expansion sizes in DM2 patients is huge. The smallest reported mutation varies between 55 and 75 [CCTG] and the largest expansions have been measured to be up to about 11,000 repeats (Liquori et al., 2001; Bachinski et al., 2009). The size of the [CCTG] repeat expansion in DM2 seems to be related, in large part, to the age of the patient and not necessarily to the severity of symptoms or manifestations as in DM1 (Day et al., 2003; Udd et al., 2003). DM2 expansion is larger than that observed in other expansion diseases. Moreover, it is the first pathogenic tetranucleotide expansion described and the degree of somatic heterogeneity of the repeat expansion is unprecedent (Liquori et al., 2001).
Usually, in other expansion disorders the amplification by polymerase chain reaction (PCR) combined with Southern Blot Analysis (SBA) are enough to confirm the presence of large expansions (Buxton et al., 1992; Harley et al., 1992; Matsuura et al., 2000) and this also applies for DM2 disease. Although the somatic heterogeneity complicates the molecular diagnosis of DM2, the combination of a long-range PCR and SBA is still the most common technique used to evaluate the expansion size.
For this reason in this study, we developed an improved molecular assay that can be used for detecting [CCTG] expansion in the molecular diagnosis of DM2. In total, 126 subjects were analyzed: 74 DM2 positive, 39 DM2 negative, and 13 DM2 negative/DM1 positive. Comparative analysis, based on the fluorescence in situ hybridization (FISH) (Cardani et al., 2004), using [CAGG]5 probe, on skeletal muscle biopsy sections, revealed that this assay correctly identified all samples. Cardiac and neuromuscular assessments were used to characterize the population and to support the new molecular assay.
An accurate genetic diagnosis of DM2 will improve care by facilitating better monitoring of the diverse clinical features known to be part of the disease, including early onset myotonia, cataracts, and cardiac and neuromuscular involvements.
Materials and Methods
Patients
From November 2013 to December 2014 a cohort of 126 patients attending the Neuromuscular Center at IRCCS Policlinico San Donato has been the subject of this study. DM2 and DM1 patients analyzed are enrolled in the Italian Myotonic Dystrophy Registry.
All patients were analyzed for the genetic determination of DM2 using the Myotonic Dystrophy type 2 kit-FL assay. All individuals were of Italian nationality. Patients were selected as follows: 35 DM2 positive and 13 DM2 negative/DM1 positive previously confirmed by molecular diagnosis (home-made assay such as: conventional PCR, short and long-range PCR), 26 DM2/DM1 negative (healthy subjects) and 52 patients with clinical diagnosis of suspected DM2. These new cases with unknown genotype were enrolled because they presented one or more DM2 features: cataracts, positive family history, myotonia, and proximal weakness. The inclusion of DM1 positive has allowed a better evaluation of the specificity.
Molecular analysis
Genomic DNA was extracted from white blood cell, using the High Pure PCR Template Preparation Kit, Roche from 114/126 subjects after written informed consent. The quality and quantity of the extracted DNAs were determined by a spectrophotometer (NanoDrop). We did not use DNA from tissue other than white blood cells. Molecular analysis of the remaining 12 subjects was performed on DNA extracted in other laboratories. The technical validation of each analytical run was subjected to internal quality control evaluation. Positive (with high or low CCTG expansions) and negative controls were patients with a previous molecular diagnosis. In addition, after the genetic test, each patient was retrospectively reviewed for the distribution of pathologic alleles containing [CCTG] repeats.
Myotonic Dystrophy type 2 kit-FL (Experteam s.r.l, Venezia, Italy)
The kit is composed of a ready-to-use Master Mix (DM Master MIX™), a long-range Polymerase (DM DNA Polymerase™), Digoxigenin-labeled probe, DNA Molecular Weight Markers VII and VIII, DIG-labeled (Roche Diagnostics), and step-by-step instructions and suggestions for optimization of the analysis.
Long-PCR amplification
To improve detection of the DM2 expansion, we developed a long-PCR combined with SBA based on previous methods by Schoser et al., 2004. The design of the primers and the type of Taq Polymerase used in this assay are different and under patent.
One microgram of genomic DNA of each patient was amplified in a reaction volume of 100 μL, containing 55 μL of DM Master MIX and 2 μL of DM DNA Polymerase. PCR conditions were: one cycle of 10 s at 96°C and 3.5 min at 95°C; 31 cycles of 30 s at 95°C, 30 s 58°C, and 3 min at 72°C +15 s increment; and finally 18 min at 68°C. The amplifications were performed by MyCycler instrument (BioRad).
Southern blot analysis
To determine the larger [CCTG] expansions, SBA of long-range PCR products was performed using nonradioactive-labeled probe. Thirty-five microliters of PCR products were separated on 1% agarose gel and transferred to a nitrocellulose filter membrane. The hybridization was carried out with 5′DIG-labeled [GGCA]6 probe at 65°C in a DIG Easy Hybridization buffer. Blots were washed twice with 2× saline sodium citrate buffer/0.1% sodium dodecyl sulfate. Then the filters were incubated with anti-digoxigenin alkaline phosphatase conjugate (Roche Diagnostics) and this one was detected by the addition of ready-to-use CDP-Star (Roche Diagnostics). The chemiluminescence signal was visualized on the ChemiDoc Instrument (BioRad) after several exposures. The bands obtained were compared with two DNA molecular weight markers (VII and VIII, respectively, Roche Diagnostics) to better detect the number [CCTG] repeats or the range of expansion.
Capillary electrophoresis
The assay was designed to produce amplified labeled with 6-FAM. The electrophoretic runs were performed and analyzed by ABI PRISM® 3100 Genetic Analyzer. The kit includes the use of the Standard GeneScan-500ROX™ (ABI P/N 401734).
Muscle histopathology and FISH analysis
Biceps brachii muscle biopsies from 52 clinically suspected DM2 patients and from 15 previously genetically confirmed DM2 patients were analyzed. Muscle tissue was fresh frozen in isopentane cooled in liquid nitrogen. Histopathological analysis was performed on serial sections (8 mm) processed for routine histological or histochemical stainings. A standard myofibrillar ATPase staining protocol was used after preincubation at pH 4.3, 4.6, and 10.4. The most typical alterations, such as nuclear clump fibers (i.e., aggregates of myonuclei with a thin rim of cytoplasm), nuclear centralization, and fiber size variability were evaluated on serial muscle sections. FISH was performed on muscle frozen sections using a [CAGG]5 probe as previously reported by Cardani et al., 2004 to verify the presence of ribonuclear inclusions.
Clinical data
Serum Creatine Kinase concentration was measured in DM2-positive patients using Roche Diagnostics immunoassays. CK was considered normal if <170 U/L in women and <200 U/L in men. All assays were performed in a blinded study according to the manufacturer's instructions by experienced laboratory personnel.
Complete clinical cardiac evaluation was carried out through physical examination, standard 12-lead ECG, and 2D echocardiography. The signal-averaged ECG was recorded during sinus rhythm. Patients were not receiving any treatment during the evaluation. Patients were identified as having mild ECG abnormalities if they had a PR interval ≥200 ms (atrioventricular block [AVB]) and a QRS duration ≥100 ms (bundle branch block [BBB]). The ECG abnormalities were considered severe if PR interval ≥240 ms, QRS duration ≥120 ms (Groh et al., 2008). Echocardiography was used to exclude significant valvulopathy and elevated right ventricular systolic pressure. Wall thickness, cavity diameters, and ejection fraction (EF) were measured on echocardiograms. Left ventricular systolic dysfunction was diagnosed when EF was lower than 50% (Lang et al., 2005). The analyses were performed without the cardiologists knowing the diagnosis and the clinical status of the patients.
Muscle strength was determined using the modified Mega Medical Research Council (MMRC) scale on 15 muscle groups on the right and on the left side, adding up to a total of 150 for normal muscle strength.
Statistical analysis
The [CCTG] expansion of each group was expressed as median and range. Overall statistical significance has been calculated by using Student's t-test. Probability values p < 0.05 were considered statistically significant.
Results
Patients
Demographic characteristic and age at the onset of DM2 patients are shown in Table 1. No significant differences were found between men and women.
Molecular analysis
Usually, the first step in molecular analysis of DM2 is to analyze whether an individual has two alleles with a low number of repeats. In contrast, if only one allele shows a low size, subsequent techniques such as SBA are used to detect possible repeat expansions. Using SBA by nonradioactive [GGCA]6 probe, expanded ZFN9/CNBP alleles can appear as single discrete bands, multiple fragments, or smears due to somatic heterogeneity. Additional discrete bands may be due to the PCR conditions and are partly dependent on the Taq polymerase used in the molecular test. In this study, we improved and validated a new molecular diagnostic test Myotonic Dystrophy type 2 kit-FL to better characterize DM2 disease. The assay is based on the combination of two steps: long-range PCR associated with SBA. A cohort of 126 individuals (56 males and 70 females) was subjected to this new genetic analysis. Out of 126 individuals tested, 74 (58.7%) were DM2 positive while 52 (41.3%) were DM2 negative (Table 2). In particular, all the 35 previously genetically confirmed DM2 patients, using several home-made assays, were correctly diagnosed as DM2 positive. Among the 52 DM2 suspected new cases with unknown genotype, 39 were DM2 positive and 13 were DM2 negative. All DM2-negative/DM1-positive patients and the healthy subjects were identified as DM2-negative patients. In addition, using this kit assay we have not observed false positive, and all negative controls (healthy subjects and DM2 negative-DM1 positive) were free of smears on the blots (Fig. 1). It should be noted that our kit assay is able to work correctly also using different DNA extraction methods. The same results were obtained by persons with different degrees and various backgrounds.
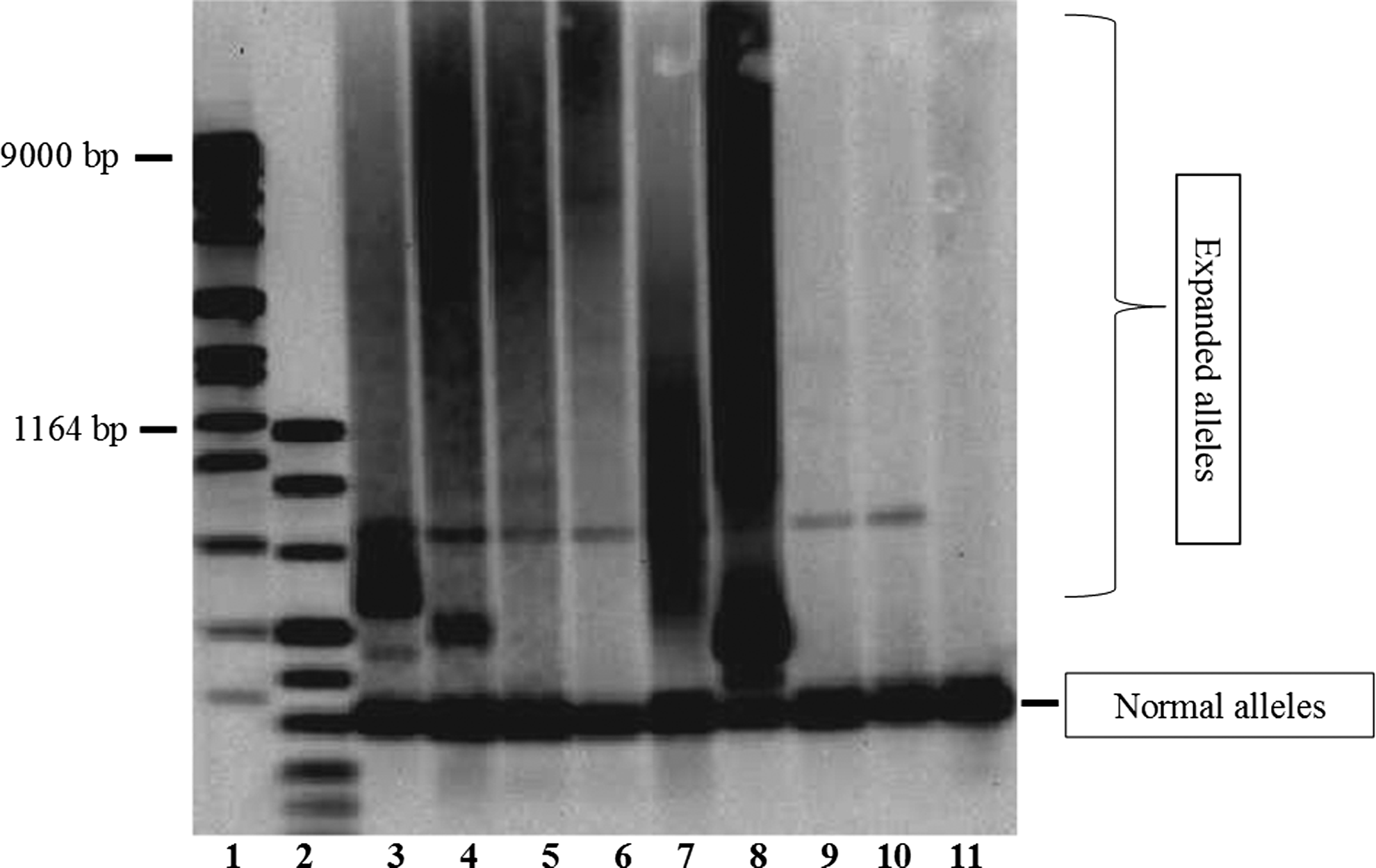
Expansions detection by Southern Blot Analysis. Lanes 1 and 2 are DNA Molecular Weight Marker (VII and VIII, respectively, Roche Diagnostics). Results for myotonic dystrophy type 2 (DM2)-affected individuals are shown in lanes 3, 4, 5, 6, 7, and 8. Results for DM2-unaffected patients are shown in lanes 9, 10, and 11 in lanes 3, 4, 5, 6, 7, and 8. Results for DM2-unaffected patients are shown in lanes 9, 10, and 11.
Number of patients resulted positive or negative/number of patients examined.
DM2, myotonic dystrophy type 2; FISH, Fluorescence in situ hybridization.
In our population, 74/126 DM2-positive patients showed expanded alleles ranging from 75 to >5000 [CCTG] repeats (median: 1000-5000 range). SBA showed that the 9.5% (7/74) of the positive samples presented single sizeable expansion and 85% (63/74) showed multiple bands or smears.
The majority of pathological alleles, about 42% (31/74) presented repeats between 1000 and 5000 [CCTG], while 35% (26/74) showed a smear above the 5000 repeats, and the 17.5% (13/74) had an expansion between 75 and 999 repeats. No significant differences were found in [CCTG] size distribution between males and females (Table 3). In addition, all PCR products were analyzed by both SBA and by the capillary electrophoresis, since the amplicon was labeled. The analysis by capillary electrophoresis demonstrated that 5.5% (4/74) patients presented an expansion in the grey-zone range between 26 and 75 [CCTG] (Fig. 2).

Fragment-length of long-range polymerase chain reaction (PCR) products of the [CCTG] repeats in the CNBP gene. Fluorescently labeled PCR products of a healthy individual (top panel) and an affected individual (bottom panel) with one normal allele and one premutated allele (55 [CCTG] repeats) were separated by capillary electrophoresis.
Muscle histopathology and FISH analysis
Histopathological analysis and FISH analysis were performed on skeletal muscle from 52 subjects with unknown genotype, but suspected to have DM2 disease and from 15 previously genetically confirmed DM2 patients. The 39/52 DM2-positive patients and the 15 DM2 presented many histological features characteristic of DM2 biopsies, with a high percentage of fibers having centrally located nuclei, severely atrophic fibers with pycnotic nuclear clumps and hypertrophic fibers. ATPase stainings showed prevalent type 2 fiber atrophy. The 13/52 DM2-negative patients showed normal muscle biopsies, confirming the validity of new molecular analysis. Ribonuclear inclusions were present in myonuclei of 39/52 DM2-positive patients and of 15 DM2 patients (Fig. 3). No ribonuclear inclusions were found in 13/52 patients who resulted negative with our molecular kit test (Table 2).
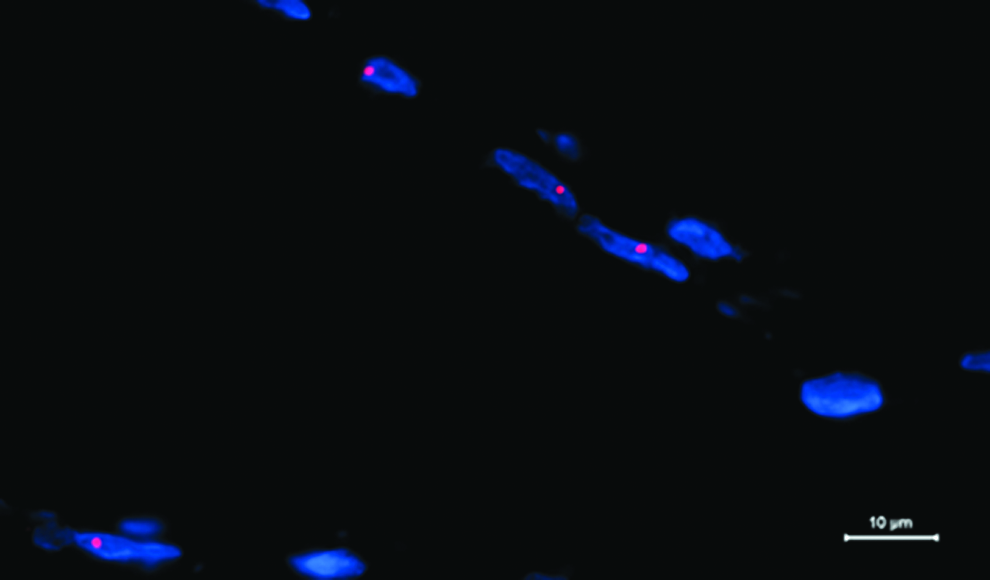
Fluorescence in situ hybridization performed on DM2 muscle section shows the accumulation of mutant RNA as ribonuclear inclusions (red spots) in nuclei (blue, DAPI).
Clinical data
Clinical data of 32/74 DM2-positive patients were used to support the genetic analysis. The results of the neuromuscular and cardiac parameters in terms of mean ± SD are reported in Table 4.
Values are reported as mean and standard deviations.
The level of serum CK was abnormally high in 87.5% (28/32) of the DM2 patients tested, with values twofold higher than the upper limits of normal range.
Cardiac characterization was available in 32/74 DM2 subjects (Table 4). Most of the 32 DM2 patients showed normal cardiac parameters. On electrocardiographic assessment, an abnormal ECG was recorded in 12/32 (37.5%) patients with PR ≥200 ms and 18/32 (56%) of the patients showed prolonged QRS duration (≥100), especially 6/18 (33%) had an incomplete BBB with QRS between 100 and 120 ms. In particular, only one DM2 male patient showed AVB. DM2 cohort showed 56% of BBB (18/32 DM2) and they were more frequent in male patients. A complete BBB was not reported on any of the female patients. The difference in PR and QRS intervals between male and female DM2 was not statistically relevant (p > 0.05). Echocardiogram data showed that most DM2 patients had normal EF. We defined an EF <50% as abnormal; on the basis of this criterion, a mitral prolapse was diagnosed in only 3/32 patients (9%), both DM2 men (EF: 35%, 40%, and 45%, respectively).
MMRC was abnormal in all DM2 patients (133 ± 14.4) and there were no differences between male and female. Myotonia was present in 28/32 (87.5%). Muscular weakness was also distinctive and proximal weakness was observed in 22/32 (69%). The posterior subcapsular iridescent cataract was present in 13/32 (41%) of our affected patients.
Discussion
The DM2 phenotype presents some similarities to DM1 such as myotonia, cataract and muscle weakness, but also some differences. The typical clinical presentation of DM2 is that of predominantly proximal muscle involvement, with muscle pain and weakness. Myotonia is variable and can be absent clinically and even on electromyography.
Both DM1 and DM2 are characterized respectively by unstable tri- or tetranucleotide repeat expansions in specific genes that lead to a spliceopathy (Brook et al., 1992; Osborne and Thornton, 2006). DNA repeat expansion mutations are dynamic and ongoing within tissues (somatic instability) and across generations (germline instability) (Pearson et al., 2005). Repeat instability probably involves the formation of unusual DNA structures during DNA replication, repair, and recombination processes that occur individually or in combination, depending on the tissue, proliferative status, and developmental stage of the cells. In DM2 disease, molecular diagnosis and interpretation of the genetic test results are complicated by the somatic instability and heterogeneity of the [CCTG] expansion.
Currently, there are different approaches for the diagnosis of the DM2 disease, but the selection of the appropriate assay depends mainly on the exact purpose of the test. Several factors may influence the decision, such as, whether the test is aimed only at proving the presence or absence of the expanded alleles or if there is a demand for exact sizing of the expanded alleles. Conventional PCR amplification represents a useful first analytic step, and if two healthy range alleles are detected, the DM2 diagnosis can be excluded. If only one allele is detected, further analysis is required. In situ hybridization is used to diagnose DM2 by determining the presence or absence of CCUG-containing RNA molecules observable as ribonuclear foci in myonuclei of muscle biopsies (Cardani et al., 2004; Sallinen et al., 2004). Nevertheless, this FISH technique is not able to diagnose patients with small expansion between 26 and 75 permutated sizes (article in preparation).
However, if sizing of the alleles is required, SBA of enzymatically digested genomic DNA and long-range PCR amplifications are still recommended and both these techniques are suitable for the detection of expanded alleles in different ranges.
In the present work we developed a powerful diagnostic ready-to-use assay based on long-range PCR and SBA for the detection of pathological alleles associated with DM2 disease. In addition, the amplification by long-range PCR with specific flanking sequences of the CNBP/ZNF9 gene and the use of a nonradioactive [CCTG]6 probe for SBA make it highly sensitive and specific. Also, the strength of this test is that PCR products are labeled and may also be analyzed by automated capillary electrophoresis, allowing it to be able to identify small expansions hard to observe by SBA. However, one disadvantage of this kit assay is due to somatic heterogeneity of DM2 repeat size, thus, in many cases expanded DM2 alleles may appear as smears or multiple fragments on SBA. In these cases, the exact sizing of the repeat expansions may be more complicated. Our results demonstrate that the specificity, sensibility, and accuracy of this new molecular test were very high (>99%), also with different methods for genomic DNA extraction. Indeed our kit assay has correctly recognized all previously genetically confirmed DM2 patients while, as expected, all the DM1-positive patients were negative. Among the patients with suspected DM2, the kit assay has been able to identify correctly the DM2-positive patients who also presented ribonuclear inclusions evidenced by FISH in muscle biopsies and a muscle histopathology suggestive of DM2.
By using this molecular test is also possible to determine a size range of the expansion. According to EMQN 2008 classification of DM2, repeats up to 26 [CCTG] units are considered to be nonpathogenic while disease-associated alleles contain 75 units and more. Alleles between 26 and 75 [CCTG] units (gray area) are very rare, however, their clinical relevance has not yet been described and characterized. Several laboratories suggest that the most frequently encountered pathogenic repeats reside in the high range (with a mean of 5000 repeats). The majority of the DM2 patients examined in this study showed pathological alleles ranging between 1000 and 5000. Clinical data reporting cardiac and neuromuscular involvement of DM2 patients examined in this work was used to support genetic diagnosis and were in line with those reported by other authors (Sansone et al., 2013).
However, while for DM1 there is a direct correlation between the size of the expansion and the severity of disease, for DM2 this association has not been reported.
Conclusions
The results of our study demonstrate that the Myotonic Dystrophy type 2 kit-FL assay appears to be a useful ready-to-use laboratory product for the DM2 diagnosis and for determining the range size of the [CCTG] expansions. Suominen et al. (2011) demonstrated that mutations for DM (DM1 and DM2) are much more prevalent than previously estimated, and that DM2 may even be the most commonly inherited muscle disease in the European populations. In particular, their results demonstrated that the vast majority of DM2 patients currently remain undiagnosed. For this reason, the new molecular diagnostic assay for DM2 disease could be very helpful to recognize DM2, so far unrecognized on clinical background, very often misdiagnosed with the more severe form of DM1 (Steinert'disease).
Therefore, the correct diagnosis of DM2 by the Myotonic Dystrophy type 2 kit-FL assay is mandatory in terms of prognosis, management, and genetic counseling (Bugiardini et al., 2014).
Footnotes
Acknowledgments
This study was financed by IRCCS Policlinico San Donato. The authors are grateful to Dr. Federica Schiavon of Experteam s.r.l (Venezia, Italy), who provided the Myotonic Dystrophy type 2 kit-FL.
Author Disclosure Statement
No competing financial interests exist.