
Abstract
Aims: Coronary artery disease (CAD), including myocardial infarction (MI), is a common complex disease caused by atherosclerosis. Although more than 50 genetic variants have been associated with CAD, these loci collectively account for only 10% of CAD cases. Genetic variants of low and rare frequencies have been proposed as the main causes of CAD. SIRT6, one of the highly conserved NAD-dependent class III deacetylases, has been implicated in cardiovascular diseases. Considering the important roles that SIRT6 plays in the cardiovascular system, inflammation, and lipid and cholesterol metabolism, genetic variants were hypothesized to contribute to MI development. Methods: The promoter regions of the SIRT6 gene were genetically analyzed in large cohorts of MI patients (n = 371) and ethnically-matched controls (n = 383). Results: A total of 15 DNA sequence variants (DSVs) were identified, including seven single-nucleotide polymorphisms (SNPs). Two novel heterozygous DSVs, g.4183823G>C and g.4183742G>A, were identified in two MI patients but in none of the controls. Two SNPs, g.4183685T>C (rs4359565) and g.4182942C>A (rs3760905), were found in MI patients with significantly higher frequencies compared with controls. Conclusions: These DSVs identified in MI patients may alter the transcriptional activity of the SIRT6 gene promoter and alter SIRT6 levels which might contribute to the risk of MI.
Introduction
C
Sirtuin 6 (SIRT6) is a member of a highly conserved family of NAD(+)-dependent deacetylases. SIRT6 has diverse functions in genomic stability, glucose and lipid metabolism, stress resistance, and life span by regulating gene expression and protein activities. Clinically, SIRT6 has been implicated in cardiovascular diseases, diabetes, obesity, inflammation, and cancer (Tennen and Chua, 2011; Etchegaray et al., 2013; Kugel and Mostoslavsky, 2014; van Meter et al., 2014). In animal experiments, mice with SIRT6 gene deletion develop normally for the first 2 weeks and die at around 1 month of age, probably due to accelerated aging process and hypoglycemia (Mostoslavsky et al., 2006). A recent study indicates that SIRT6 controls embryonic stem cell fate in mice (Etchegaray et al., 2015).
Human studies and animal experiments have shown that SIRT6 provides protective effects in cardiovascular diseases, including cardiac hypertrophy, ischemia-reperfusion, and heart failure (Winnik et al., 2015). SIRT6 blocks the development of cardiac hypertrophy by directly regulating insulin growth factor (IGF)-Akt signaling (Sundaresan et al., 2012; Pillai et al., 2014). Akt signaling is known to regulate cardiac growth, contractile function, and coronary angiogenesis (Shiojima and Walsh, 2006). In model animals, SIRT6 mediates the protective role of nicotinamide mononucleotide adenylyltransferase from hypertrophy and suppresses cardiomyocyte hypertrophy via inhibition of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) (Cai et al., 2012; Yu et al., 2013). During ischemia-reperfusion, SIRT6 protects the heart from apoptosis (Cattelan et al., 2015). Moreover, overexpressed SIRT6 protects cardiomyocytes from hypoxic damage (Maksin-Matveev et al., 2015). SIRT6 also inhibits cardiac fibroblast differentiation into myofibroblasts (Tian et al., 2015).
SIRT6 has been shown to play important roles in vascular system, lipid metabolism, cholesterol biosynthesis, and inflammation. In human endothelial cells, SIRT6 confers protection from telomere and genomic DNA damage (Cardus et al., 2013). SIRT6 regulates the differentiation of vascular smooth muscle cell in response to the cyclic strain (Yao et al., 2014). SIRT6 deficiency increases the expression of genes for triglyceride synthesis and triglyceride levels (Kim et al., 2010; Yang et al., 2011). SIRT6 gene overexpression reduces fat accumulation and decreases LDL-cholesterol and triglyceride levels (Kanfi et al., 2010; Tao et al., 2013a). SIRT6 promotes the secretion of tumor necrosis factor-α (TNF-α) and inhibits NF-κB, a key transcription factor pathway for many key biological processes (Jiang et al., 2013; Maksin-Matveev et al., 2015). SIRT6 reduces inflammatory response by blocking the NF-κB pathway, and SIRT6 deficiency activates NF-κB signaling (Kawahara et al., 2009; Lee et al., 2013).
Accumulating evidence has revealed the beneficial effects of SIRT6 on cardiovascular system, lipid metabolism, and inflammation. Thus, we speculated that altered expression of SIRT6 gene, caused by the DNA sequence variants (DSVs) with its regulatory regions, may contribute to the CAD development. In this study, we genetically analyzed the SIRT6 gene promoter in large cohorts of MI patients and healthy controls.
Materials and Methods
Study subjects
All MI patients (n = 371; age 24-85 years, mean age 61.66 years; males 276 and females 95) were recruited from the Division of Cardiology, Affiliated Hospital of Jining Medical University, Jining Medical University, Jining, Shandong, China. MI diagnosis was based on clinical symptoms, abnormal electrocardiogram, and elevated levels of plasma cardiac necrosis markers. Ethnic-matched healthy controls (n = 383; age 22-79 years, mean age 48.06 years; males 207 and females 176) were recruited from the Physical Examination Center in the same hospital. The controls with familiar CAD histories were excluded from the study. This study was approved by the Human Ethics Committee of the Affiliated Hospital of Jining Medical University. Informed consents were obtained from patients and guardians.
Genetic analysis
Peripheral venous blood was collected, and leukocytes were isolated. Genomic DNAs were extracted from leukocytes with DNeasy Blood and Tissue Kit (QIAGEN, Valencia, CA). The promoter region of SIRT6 gene, from −1307 bp upstream to +55 bp downstream to the transcription start site, was generated by polymerase chain reaction (PCR) and analyzed by direct sequencing. Genomic DNAs (100 ng) were used as PCR templates. PCR primers were designed with the genomic sequence of human SIRT6 gene (GenBank accession number, NC_000019.10). Two overlapped DNA fragments covering the SIRT6 gene promoter region, −1307 to −694 bp (614 bp) and −756 to +55 bp (811 bp), were generated with the PCR primers in Table 1. Direct DNA sequencing was performed with 3730 DNA Analyzer (Applied Biosystems, Foster City, CA). For heterozygous deletion and insertion DSVs, the DNA fragments were first subcloned into T vector and then directly sequenced. DNA sequences were aligned and compared with wild-type SIRT6 gene promoter, and DSVs were identified. Distributions of DSVs were compared between MI patients and controls using SPSS v13.0. p < 0.05 was considered statistically significant.
PCR primers are designed based on the genomic DNA sequence of the SIRT6 gene (NC_000019.10). The transcription start site is at the position of 4182599 (+1).
PCR, polymerase chain reaction.
Results
In total, 15 DSVs were identified in this study population, including seven single-nucleotide polymorphisms (SNPs), six novel heterozygous DSVs, one heterozygous insertion DSV, and one heterozygous deletion DSV. Distributions and locations of the DSVs within the SIRT6 gene promoter region in MI patients and controls are summarized in Table 2 and Figure 1. Two novel heterozygous DSVs, g.4183823G>C and g.4183742G>A, were identified in two MI patients but in none of the controls. Two SNPs, g.4183685T>C (rs4359565) and g.4182942C>A (rs3760905), were found in MI patients with higher frequencies compared to controls (p = 0.032 and p = 0.007, respectively). One SNP, g.4183674C>T (rs183682646), and four novel heterozygous DSVs, g.4183280T>A, g.4183208C>T, g.4183202T>C, and g.4182791del, were only found in controls. The other two novel DSVs, g.4183836_37ins and g.4182991C>A, and four SNPs, g.4183272C>T (rs200490859), g.4183158A>C (rs3760907), g.4183126G>A (rs186636683), and g.4183047G>C (rs3760906), were found in both MI patients and controls with similar frequencies (p > 0.05). All the DSVs identified in MI patients and controls are depicted (Fig. 2).

Locations of the DNA sequence variants (DSVs) within the SIRT6 gene promoter in myocardial infarction (MI) patients and controls. The numbers represent the SIRT6 genomic DNA sequences (GenBank accession number NC_000019.10). The transcription start site is at the position 4182599 in the first exon.
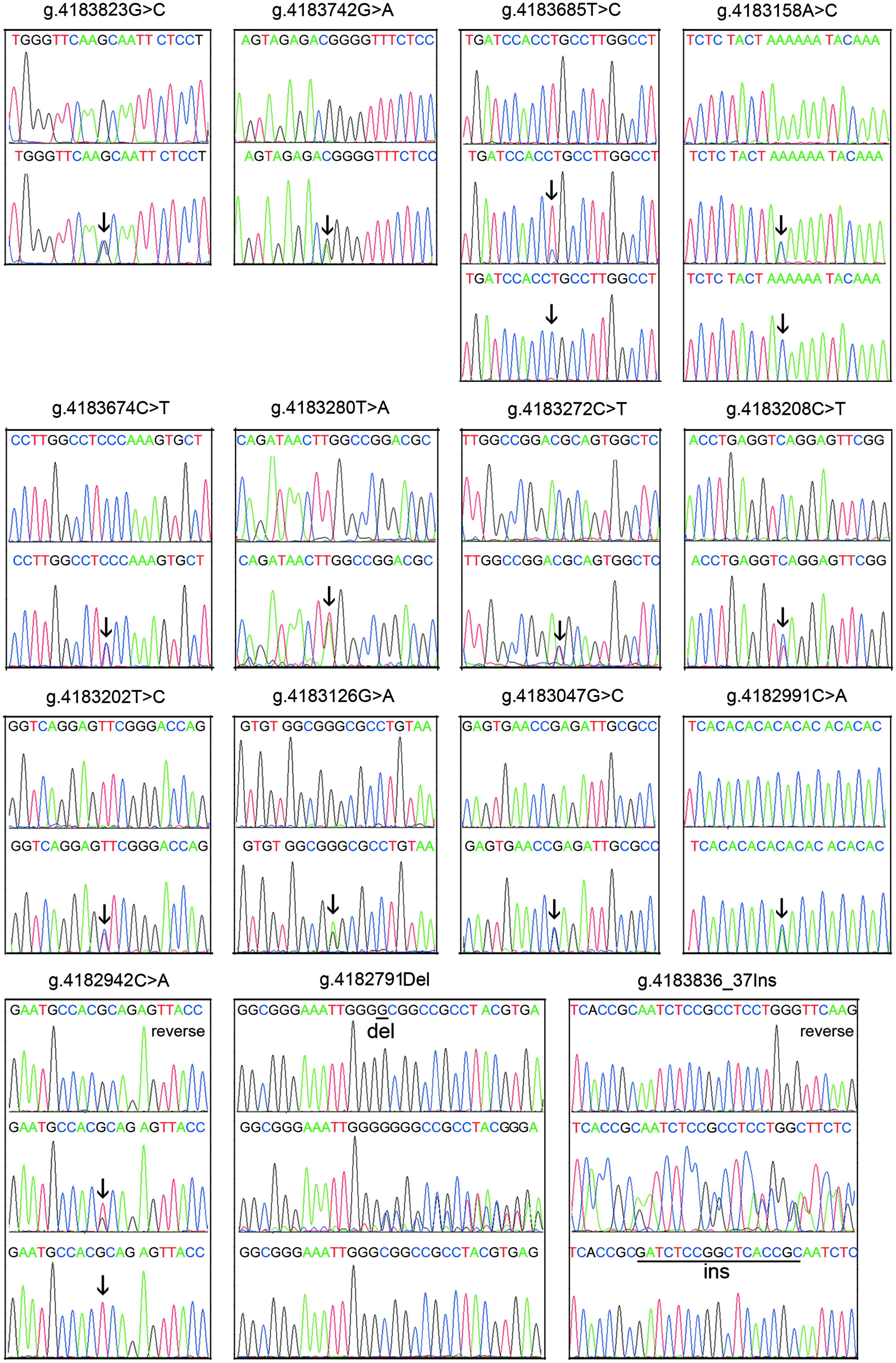
A schematic representation of the DSVs within the SIRT6 gene promoter. All the DSVs identified in MI patients and controls are depicted. All the DSVs, except marked with reverse, are shown in forward orientations. For DSVs g.4183823G>C, g.4183742G>A, g.4183674C>T (rs183682646), g.4183280T>A, g.4183272C>T (rs200490859), g.4183208C>T, g.4183202T>C, g.4183126G>A (rs186636683), g.4183047G>C (rs3760906), and g.4182991C>A, top and bottom panels show wild-type and heterozygous DSVs, respectively, which are marked with arrows. For DSVs g.4183836_37ins and g.4182791del, top, middle, and bottom panels show wild-type, heterozygous, and cloning DNA sequences, respectively. The deletion and insertion are underlined and labeled. Color images available online at www.liebertpub.com/gtmb
DSVs are located upstream (-) to the transcription start site of SIRT6 gene at 4182599 of NC_000019.10.
DSV, DNA sequence variant.
Analysis of SIRT6 gene promoter regions with JASPAR program (http://jaspar.genereg.net) suggested that the DSVs identified in MI patients may change the binding sites for putative transcription factors. The DSV g.4183823G>C may abolish a signal transducer and activator of transcription 1 (STAT1) site and create sites for breast cancer 1, early onset (BRCA1) and homeobox protein A5 (HOXA5). The DSV g.4183742G>A may abolish the sites for transcription factors SP1 and REL (NF-κB P65 subunit) and create a sex-determining region Y-box 10 (SOX10) site. The SNP g.4183685T>C (rs4359565) may abolish an upstream transcription factor 2 (USF2) and a bHLH-Zip protein MAX binding sites and create sites for transcription factors SP2, E2F1, E2F3, E2F4, and E2F6. The SNP g.4182942C>A (rs3760905) may abolish a site for a heterodimer of hypoxia-inducible factor 1A (HIF-1A) and aryl hydrocarbon receptor nuclear translocator (ARNT).
Furthermore, the DSVs identified in controls were also analyzed with JASPAR program. The SNP g.4183674C>T (rs183682646) may create the sites for nuclear factor 1 C-type (NFIC) and helicase-like transcription factor (HLTF). The DSV g.4183280T>A may abolish sites for nuclear factor 1 X-type (NFIX), NFIC, and HLTF and create the sites for homeobox transcription factor 1 (HMBOX1) and transcription factor LBX1. The DSV g.4183208C>T may abolish nuclear receptor subfamily 2, group F, member 1 (NR2F1), CAMP-responsive element-binding protein 1 (CREB1), and nuclear receptor subfamily 4, group A, member 2 (NR4A2). The DSVs g.4183202T>C and g.4182791del do not affect any binding sites.
In addition, the DSVs identified in both MI patients and controls were analyzed with JASPAR program. The DSV g.4183836_37ins may abolish the site for transcription factor SP1 and create a site for Rhox homeobox family, member 1 (RHOXF1). The DSV g.4182991C>A may create sites for forkhead transcription factors D2 (FOXD2), FOXL1, and FOXP3. The SNP g.4183272C>T (rs200490859) may create binding sites for transcription factors ETS1, ETS translocation variant 1, (ETV1), ETV4, and ETV5. The SNPs g.4183158A>C (rs3760907), g.4183126G>A (rs186636683), and g.4183047G>C (rs3760906) do not change any binding sites for known transcription factors.
Taken together, the DSVs identified in AMI patients may alter the transcriptional activity of the SIRT6 gene promoter. Biological functions of these DSVs will be investigated with dual-leuciferase reporter gene assays in our future studies.
Discussion
To date, few studies on genetic analysis of the SIRT6 gene have been reported in human diseases. In a group of stroke-free subjects, genetic variants of SIRT6 gene have been associated with carotid atherosclerotic plaque (Dong et al., 2011). In an aged population, SIRT6 gene SNP g.4176088T>C (rs107251) has been associated with a decrease in life span (TenNapel et al., 2014). In MI patients, we found two novel DSVs, g.4183823G>C and g.4183742G>A, and two SNPs with significant higher frequencies, g.4183685T>C (rs4359565) and g.4182942C>A (rs3760905), within the SIRT6 gene promoter. These DSVs may change SIRT6 gene expression level, contributing to the MI development. Therefore, our study linked genetic variants of the SIRT6 gene with the common human disease, CAD.
The human SIRT6 gene is localized to chromosome 19p13.3 (Frye, 2000). The promoter region of the human SIRT6 gene has not been characterized in detail. In human cells, the SIRT6 gene is regulated by Runt-related transcription factor 2 (RUNX2), miR-34a, and oxygen and glucose deprivation (Lee et al., 2013; Lefort et al., 2013; Choe et al., 2015). RUNX2 is a member of the RUNX family of transcription factors with a Runt DNA-binding domain, which is required for oxidized low-density lipoprotein-induced gene expression in vascular smooth muscle cells (Farrokhi et al., 2015). In diabetic atherosclerotic plaques, SIRT6 levels are significantly reduced compared to nondiabetic plaques (Balestrieri et al., 2015). In diverse types of cancers and tumors, alterations of SIRT6 levels have also been observed (Sebastián et al., 2012; Marquardt et al., 2013; Ming et al., 2014; Wu et al., 2014). In this study, the DSVs identified within the SIRT6 gene promoter may change SIRT6 gene expression levels. Therefore, dysregulation of SIRT6 gene expression may contribute to the human diseases.
SIRT6 is localized in the nucleus and acts as both an ADP-ribosylase and a deacetylase. SIRT6 deacetylates histone H3 at lysines 9 and 56, which is required for the expression and regulation of a variety of genes (Michishita et al., 2008, 2009; Etchegaray et al., 2013). A number of downstream targets and partners of SIRT6 have been identified. For example, SIRT6 directly interacts with c-Jun and binds to the promoters of IGF signaling-related genes, attenuating IGF-Akt signaling (Sundaresan et al., 2012). By partnering with forkhead box transcription factor 3 (FOXO3), SIRT6 represses the expression of sterol regulatory element-binding protein 1 (SREBP), SREBP2, and proprotein convertase subtilisin/kexin type 9 (PCSK9) gene and their target genes, which are crucial in regulating low-density lipoprotein-cholesterol (LDL-C) homeostasis (Elhanati et al., 2013; Tao et al., 2013a, 2013b). SIRT6 interacts with NF-κB subunit to regulate NF-κB-targeting gene expression (Kawahara et al., 2009; Yu et al., 2013; Tian et al., 2015). SIRT6 also controls HIF1-α-regulated genes in glucose metabolism (Zhong et al., 2010). SIRT6 gene overexpression induces autophagy via attenuation of IGF-Akt-mTOR signaling, and SIRT6 knockdown inhibits autophagy (Takasaka et al., 2014). Autophagy, a conserved cellular process for digestion of long-lived proteins and organelles, has been associated with cardiovascular diseases (Gatica et al., 2015). In human umbilical vein endothelial cells, SIRT6 loss leads to upregulation of genes involved in inflammation, vascular remodeling, and angiogenesis (Lappas, 2012). Collectively, changed SIRT6 levels may affect its targets and partners and contribute to the MI development through different pathways, including cardiovascular function, lipid metabolism, and inflammation.
In conclusion, we genetically analyzed the SIRT6 gene promoter in MI patients. These DSVs identified in MI patients, including SNPs, may alter transcriptional activity of SIRT6 gene promoter and change SIRT6 level, contributing to the MI development. The related molecular mechanisms are being investigated in our laboratory. The findings may deepen our understanding of genetic causes of MI and provide a genetic basis for potential personalized therapy for MI patients.
Footnotes
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 81370271) and Taishan Scholar Program of Shandong, China.
Author Disclosure Statement
No competing financial interests exist.