
Abstract
Background: Familial exudative vitreoretinopathy (FEVR, OMIM 133780) is a severe inherited retinal disorder characterized by incomplete retinal vascular development and neovascularization. At least five genes have been reported to be associated with FEVR, including NDP, LRP5, FZD4, TSPAN12, and ZNF408. Recently reported data showed that mutations in the KIF11 gene can also lead to FEVR conditions. Previous studies suggested that known mutations only explain approximately 40-60% of FEVR cases in different populations. Purpose: To investigate the causative genetic mutations in four Indian families with FEVR. Methods: Whole exome sequencing was carried out to analyze the genomic DNA samples from the four FEVR proband patients and Sanger sequencing was utilized to verify all identified polymorphisms. A luciferase assay was used to test the mutant protein activity. Results: We identified four novel LRP5 missense mutations in these FEVR families: c.C1042T (p.R348W), c.G1141A (p.D381N), c.C1870T (p.R624W), and c.A4550G (p.Y1517C). The luciferase assay demonstrated that all four of these LRP5 mutations led to significant reduction of enzymatic activity with response to NORRIN, suggesting that they are pathogenic. Conclusion: Our findings expand the mutational spectrum of FEVR in the Indian population and provide some guidelines in clinical diagnosis.
Introduction
F
FEVR is a clinically heterogeneous disease: the clinical appearance of FEVR can differ greatly among patients, from completely blind to asymptomatic, even for patients from the same family or between the two eyes of an individual patient (Yang et al., 2011). FEVR is also genetically heterogeneous, to date, at least five genes, including NDP (Norrie disease protein), LRP5 (low-density lipoprotein receptor-related protein 5), FZD4 (frizzled class receptor 4), TSPAN12 (tetraspanin 12) and ZNF408 (zinc finger protein 408) have been reported to associate with FEVR and can explain about half of FEVR cases (Collin et al., 2013). Recently reported data showed that mutations in KIF11 also lead to FEVR conditions (Robitaille et al., 2014). FEVR can be inherited in an autosomal dominant, recessive, or X-linked pattern (Gow and Oliver, 1971; Feldman et al., 1983; Plager et al., 1992; Chen et al., 1993; Shastry and Trese, 1997; Crecchio et al., 1998). The proteins encoded by the FEVR genes, NPD, FZD4, LRP5, and TSPAN12, are involved in the Norrin/β-catenin signaling pathway, which play important roles in most embryonic developmental processes, including cell-fate determination, proliferation, motility, and the establishment of the primary axis of the body during vertebrate embryogenesis (Wodarz and Nusse, 1998; Logan and Nusse, 2004). The inactivation of Norrin/β-catenin signaling pathway may cause incomplete eye or retinal development (Xu et al., 2004; Liu et al., 2006; Osakada et al., 2007).
In this study, we carried out the whole exome sequencing (WES) analysis and identified four novel LRP5 mutations in patients with FEVR from Indian families. Further functional studies confirmed that the proteins harboring identified mutations were dysfunctional and failed to activate Norrin/β-catenin signaling pathway by luciferase assay, thus implicating that these mutations are pathologic. This research revealed the genetic causes of FEVR in corresponding families and expanded the mutation spectrum of FEVR in the Indian population.
Materials and Methods
Patient recruitment and ethics statement
This study was approved by the Institutional Ethics Review Board of Aravind Medical Research Foundation of India and the Hospital of the University of Electronic Science and Technology of China and the Sichuan Provincial People's Hospital, and all experiments were carried out in accordance with the approved study protocols. For this study, patients and 200 control individuals without eye diseases were recruited from the Aravind Eye Hospital of India, and written informed consent was obtained from all individuals who participated in this study or from their legal guardians in the case of minors. All participants were evaluated by a clinical ophthalmologist based on fundus photographic and angiographic changes. The angiographic examinations were assessed by intravenous injection of fluorescein dye. Inclusion criteria for the patients were incomplete retinal development (avascular peripheral retinal zone) and neovascularization. All the patients in our study were excluded from premature birth, drug intake by mother during pregnancy, and other ocular diseases associated with osteopenia, osteoporosis, or other defects.
Clinical information
Clinical information for the nine patients and their family members from the four Indian families who participated in this study is shown in Table 1.
F and M in the “age/sex” column represent female and male, respectively; A and UA in the last column in this table represent affected and unaffected, respectively.
Mutation screening by WES
Peripheral blood from patients with FEVR and other members of their families as well as the 200 normal control individuals was collected in EDTA anticoagulant tubes and then genomic DNA was isolated using the DNA Extraction Kits according to the protocol provided by the manufacturer (TIANGEN). DNA was stored at −20°C for use in subsequent analyses. DNA samples from the four probands (A001, B001, C001, and D001) were subjected to WES analysis at Axeq Technology, Inc., Seoul, Korea. The genomic DNA fragments containing coding regions were captured by the Agilent SureSelect Target Enrichment System (Agilent Technologies). Then, the exome-enriched DNA fragments were sequenced on the Illumina HiSeq2000sequencer (Illumina, Inc.). The sequencing reads were aligned to the reference human genome (hg19 UCSC assembly, http://genome.ucsc.edu/) using Burrows-Wheeler Aligner (BWA). Single-nucleotide variants (SNVs) and insertions or deletions (InDels) were identified by SAMtools. The following bioinformatics pipeline was applied to detect the causative variant: (1) exclusion of the low-quality variations (quality score <20; sequencing depth <4) and variants in noncoding regions or synonymous variants without affecting splicing; (2) exclusion of variants with minor allele frequency >0.01 determined with dbSNP137 (www.ncbi.nlm.nih.gov/projects/SNP/), 1000 genomes (http://browser.1000genomes.org/index.html); (3) exclusion of variants predicted to be benign by online tools (SIFT/PROVEAN, PolyPhen-2, and PMut).
Mutation confirmation by Sanger sequencing
Sanger sequencing was carried out to confirm the mutations identified by WES. PCR primers were designed and synthesized by Sangon Biotech to amplify genomic DNA fragments containing the candidate loci. The PCR products were purified by FastAP Thermosensitive Alkaline Phosphatase (Thermo Scientific Fermentas), and then were directly sequenced using BigDye version 3.1 and an ABI 3730 automated sequencer (Applied Biosystems) according to the manufacturer's instructions. DNA samples from the probands and other family members shown in Table 1 were sequenced by Sanger sequencing for the corresponding mutation. Two hundred control individuals were sequenced by Sanger sequencing for all four mutations.
Construction of expression plasmids
The cDNA encoding wild-type LRP5 (OriGene) was subcloned in-frame into the pRK5 vector (BD Bioscience) using the XbaI and HindIII sites. All mutations were introduced into the wild-type LRP5 cDNA by site-directed mutagenesis using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies). FZD4 and NORRIN expression plasmids (generously provided by Dr. Jeremy Nathans of the Johns Hopkins University) have been described previously (Xu et al., 2004). The recombinant plasmids containing pRK5-LRP5 fusion constructs were first verified by direct DNA sequencing, and then prepared for transfection using the Qiagen Plasmid Maxi Preparation Kit (Qiagen).
Luciferase assays
HEK293 cells stably harboring the Wnt/β-catenin reporter SuperTOPFlash (HEK293STF) were seeded into a 24-well plate and then cotransfected with 200 ng of NORRIN, 200 ng of FZD4, 240 ng of LRP5 (wild type or mutant), and 100 ng of pSV-β-Galactosidase Control Vector (Promega) for each well using the Lipofectamine™ 2000 Transfection Reagent (Invitrogen) according to the manufacturer's instructions. The transfected cells were used for luciferase activity assay using the Promega Luciferase Reporter Assay System after 48 h. The firefly luciferase activity was normalized to the coexpressed β-galactosidase activity. Each assay was performed in triplicate at the same time and repeated three times.
Expression of LRP5 plasmids in cells
Western blotting method was carried out to assess the expression of LRP5 plasmids. HEK293STF cells were cultured in DMEM with high glucose (Gibco) supplemented with 10% fetal bovine serum and 1% (v/v) penicillin/streptomycin at 37°C in a 5% CO2 atmosphere. For western blotting assay, HEK293 STF cells were seeded into six-well plates and then 3000 ng of LRP5 plasmid (wild-type or mutant) or 3000 ng of pCMV6 vector was transfected using the Lipofectamine™ 2000 Transfection Reagent (Invitrogen) for each well, respectively. Forty-eight hours later, the cell lysates were used for western blotting assay using LRP5 antibody (Catalog number 5731S; Cell Signaling Technology, Inc.).
Results
Identification of causative mutations
To identify the underlying genomic mutations that cause FEVR in these patients, the four probands were subjected to WES analysis. As results, we did not find any mutations or compound mutations in the known FEVR genes: NDP, FZD4, TSPAN12, and ZNF408. However, we identified four missense mutations in the LRP5 gene, which was known to be associated with FEVR. Sanger sequencing results (shown in Fig. 1) confirmed the WES results and demonstrated that these mutations cosegregate with the disease phenotype in these families. WES data output statistics are shown in Supplementary Tables S1 and S2 (Supplementary Data are available online at www.liebertpub.com/gtmb).

Pedigrees of the four FEVR families with LRP5 mutations and corresponding sequence chromatograms.
Specifically, in pedigree A, we identified a heterozygous mutation c.C1042T in LRP5 in the proband, and Sanger sequencing revealed that his affected father and sister carried the same mutation. This mutation led to the replacement of an arginine by a tryptophan at codon 348 (p.R348W), which involved a highly evolutionarily conserved residue from Homo sapiens to Xenopus tropicalis (Fig. 2). In pedigree B, we found that the proband and his affected father both carried a heterozygous mutation, c.1141G>A in exon 6. This mutation resulted in the replacement of an aspartate by an asparagine at codon 381 (p.D381N), which was also highly evolutionarily conserved. In pedigree C, a homozygous mutation in exon 9 of LRP5, c.1870C>T was identified in the proband and his affected mother, whereas the other asymptomatic members (the proband's uncle and aunty) from this family were heterozygous. This mutation causes the replacement of an arginine by a tryptophan at codon 624 (p.R624W). We also identified a missense mutation c.4550A>G in LRP5 in the proband of pedigree D, and the same mutation was found in her affected mother. This mutation leads to the change of a tyrosine to cysteine at codon 1517 (p.Y1517C), which is extremely evolutionarily conserved from H. sapiens to X. tropicalis (Fig. 2). The causative mutations in these families are shown in Table 2. SIFT and PROVEAN analyses predicted all of these mutations to be damaging, and none of these mutations were found in the 200 normal control individuals, thus emphasizing their pathogenicity.

Protein sequence alignment of human LRP5 with its orthologues. The orthologues are from the following species: Homo sapiens (NP_002326.2), Pan troglodytes (XP_508605.3), Canis lupus familiaris (XP_003432463.1), Bos taurus (XP_002699451.1), Macaca mulata (M. musculus) (NP_032539.2), Rattus norvegicus (NP_001099791.2), Gallus gallus (NP_001012915.1), Danio rerio (NP_001170929.1), and Xenopus tropicalis (NP_001093730.1).
Defective luciferase reporter activity mediated by mutant LRP5 proteins
To confirm the pathogenicity of these mutations, we conducted a luciferase assay in STF cells (described in “Materials and Methods” section). The results indicated that all four LRP5 mutations failed to induce luciferase reporter activity compared with wild-type LRP5. To be precise, mutations R348W, D381N, and Y1517C (separately) inactivated the protein. Mutation R624W still retained some residual activity compared with the wild type (Fig. 3).

Luciferase assays with the STF cell line transfected with the indicated plasmids. STF cells cotransfected with pRK5-LRP5, pCMV6-FZD4, and pCMV6-NORRIN, and assayed for luciferase reporter activity. Luciferase assay were performed in triplicate, and the results are shown as an average of three measurements.
To ascertain the expression of the four mutant LRP5 proteins in transfected cells, we carried out western blotting analysis. Our result showed that expression levels of R348W, D381N, and R624W mutant proteins were comparable with that of the wild-type protein, whereas the Y1517C mutant protein was undetectable (Fig. 4). RT-PCR result indicated that Y1517C mutant plasmid was successfully transfected and expressed (shown in Supplementary Fig S1), thus proving that the Y1517C mutant protein was unstable or cannot be detected by the LRP5 antibody used in this study.
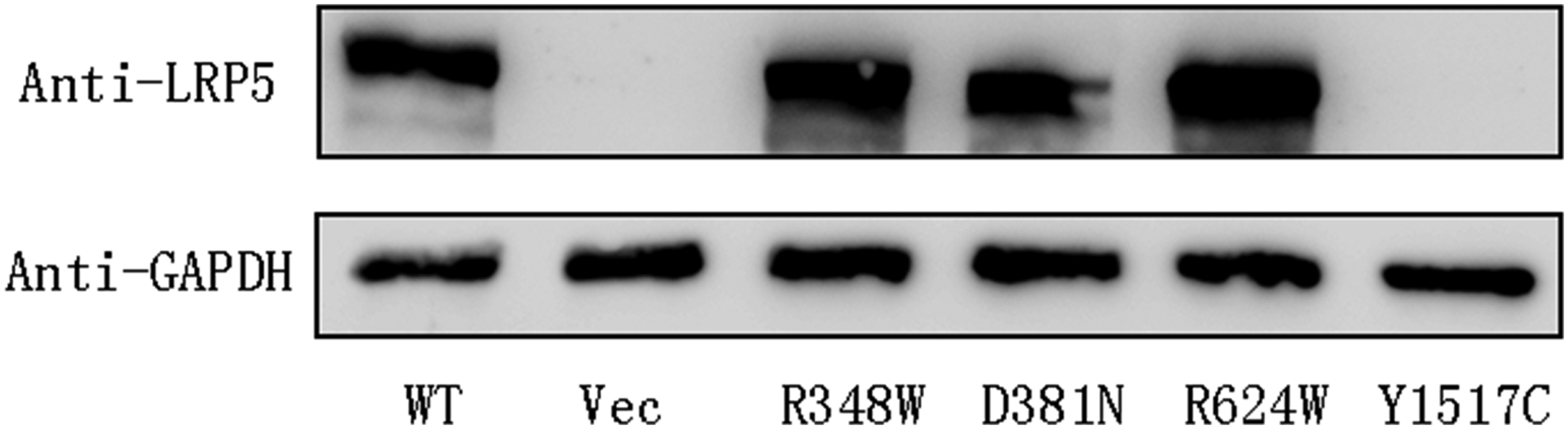
Western blot analysis by SDS-PAGE of the LRP5 mutants. Total proteins isolated from cell lysates were mixed with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer and subjected to SDS-PAGE and western blot analysis using anti-LRP5 antibody to detect LRP5 expression. GAPDH was used as the loading control and the vector plasmid (Vec) was used as negative control. The expression level of R348W, D381N, and R624W are compatible with the wild-type protein.
Discussion
The pathological features of FEVR can be manifested in different manners. Avascular peripheral retinal zone is the hallmark of the disease and in many mildly affected patients it may be the only manifestation (Shukla et al., 2003). In more severe cases, retinal complications, such as the formation of holes, folders and tears, neovascularization, exudation, vitreous hemorrhage, macular ectopia, and retinal detachment may develop, and eventually result in blindness (Nikopoulos et al., 2010). To date, there are no effective ways to treat FEVR, thus implicating the importance of prenatal diagnosis, especially for those families with FEVR histories. WES analysis provides us a powerful tool to diagnose FEVR, as it is quick, accurate, and relatively low in cost. In our study, to find out the pathogenic mutations causing FEVR in the four Indian families, we subjected the DNA samples of the four probands to WES analysis. We identified four novel LRP5 mutations and demonstrated the effect of these four mutations on LRP5 activity. Our study proved that WES analysis is indeed a useful tool for clinical diagnosis.
LRP5 gene is localized on human chromosome 11q13 and encodes the 1615 amino acid single-pass transmembrane protein LRP5 (Hussain et al., 1999), which belongs to the low-density lipoprotein (LDL) receptor family. LRP5 can act with FZD and WNT to form the WNT-FZD-LRP5 complex that triggers canonical Wnt/β-catenin or the NORRIN/β-catenin signaling pathway and induce the transcription of target genes subsequently (Pinson et al., 2000; He et al., 2004). WNT/β-catenin signaling pathway is evolutionarily highly conserved and plays an important role in eye development and angiogenesis (Nikopoulos et al., 2010). Mutations in LRP5 have been proved to cause FEVR (Jiao et al., 2004). To date, more than thirty different mutations of LRP5 have been reported to relate to FEVR, including premature stop codons, missense changes, and changes that affect splicing (Fei et al., 2014). LRP5 protein consists of extracellular, transmembrane, and cytoplasmic regions. The extracellular region consists of four β-propellers, each of which is composed of six segments and followed by an epidermal growth factor (EGF)-like domain (Nikopoulos et al., 2010). The first two β-propellers were suggested to be important for interaction with WNT and FZD (Mao et al., 2001). Between the four propellers and the transmembrane domain there are three LDL-receptor-like ligand-binding domains, which range from the 1255th to 1370th amino acid. The cytoplasmic region contains one or multiple short signals for receptor internalization through coated pits.
In our study, the first two mutations (p.R348W and p.D381N) are located in the second β-propeller at a well-conserved position and result in loss of their ability to activate the FZD4/Norrin signaling. This is consistent with the effect of the previously reported mutations (p.A422T, p.L540P, and p.R570Q), which are also located at the second β-propeller (Ai et al., 2005; Fei et al., 2014). Mutation p. R624W is located at the second EGF-like domain at a not very conserved residue, but the mutant protein still lost its 85% activity compared with that of the wild type. This may be due to the replacement of a nonpolar amino acid (tryptophan) by a positively charged amino acid (arginine), changing the polarity and stability of the domain, and leading to the partial loss of the protein function. This may explain the fact that the proband and his father with homozygous mutation showed FEVR symptoms, whereas his uncle and aunt with heterozygous mutation did not display apparent vision loss. The mutation p.Y1517C is located in the cytoplasmic pro-rich domain, which plays an important role in signal transduction and protein stability. Our data showed that LRP5 protein with this mutation almost completely lost its activity, thus confirming the pathogenicity of this mutation. LRP5 mutations associated with FEVR are scattered through the gene (Nikopoulos et al., 2010). These four mutations identified in our article are not limited in a specific region of LRP5 gene, but distributed in different exons (exon 6, 9, 19). These demonstrated that all domains of LRP5 protein are essential for the signal transduction.
In conclusion, through WES analysis and functional studies, we identified four novel pathogenic LRP5 mutations in four Indian FEVR families. These findings provide additional evidence that mutations in LRP5 can cause FEVR and expand the mutation spectrum of FEVR in the Indian population, and are thus of considerable diagnostic significance.
Footnotes
Acknowledgments
The authors thank all of the patients and family members for their participation. This study was supported by grants from the National Natural Science Foundation of China (81170883 and 81430008 to Z.Y., 81271007 and 81470668 to X.Z., 81100693 to C.Q., and 81300802 to L.H.). It was also supported by grants from the Department of Science and Technology of Sichuan Province (2012SZ0219, 2014SZ0169, 2015SZ0052 [Z.Y.], 2015SZ0060 [Y.L.], 2014JQ0023, 2014FZ0122, 16CXTD0066 [X.Z.], and 2105JZ0004 [C.Q.]) and the Department of Health of Sichuan Province (130145 to X.Z.).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.