
Abstract
Objective:
Long noncoding RNAs (lncRNAs) are becoming promising biomarker candidates in various diseases as assessed via sequencing technologies. Sepsis is a life-threatening disease without ideal biomarkers. The aim of this study was to investigate the expression profile of lncRNAs in the peripheral blood of sepsis patients and to find potential biomarkers of sepsis.
Methods:
A lncRNA expression profile was performed using peripheral blood from three sepsis patients and three healthy volunteers using microarray screening. The differentially expressed lncRNAs were validated by real-time quantitative polymerase chain reaction (qRT-PCR) in a further set of 22 sepsis patients and 22 healthy volunteers.
Results:
Among 1316 differentially expressed lncRNAs, 771 were downregulated and 545 were upregulated. Results of the qRT-PCR were consistent with the microarray data. lncRNA ENST00000452391.1, uc001vji.1, and uc021zxw.1 were significantly differentially expressed between sepsis patients and healthy volunteers. Moreover, lncRNA ENST00000504301.1 and ENST00000452391.1 were significantly differentially expressed between sepsis survivors and nonsurvivors.
Conclusion:
The lncRNA expression profile in the peripheral blood of sepsis patients significantly differed from that of healthy volunteers. Circulating lncRNAs may be good candidates for sepsis biomarkers.
Introduction
S
Long noncoding RNAs (lncRNAs) are a class of noncoding RNAs that contains more than 200 nucleotides without open reading frames. lncRNAs were previously thought to be conservative and functionless but, over the past decade, important roles of lncRNAs have been discovered. lncRNAs can regulate gene and genome activity by regulating histone modification and chromatin remodeling, modulating DNA methylation, interacting with transcription factors, looping enhancers, and modifying at the posttranscriptional level (Jalali et al., 2013; Lopez-Pajares, 2016; Schmitz et al., 2016). With the advent of sequencing technologies, different lncRNA expression profiles have been observed in various diseases. Those differentially expressed lncRNAs can stably exist in blood through membrane vesicles such as exosomes and microvesicles and, therefore, become promising biomarkers for the diagnosis and prognosis of diseases (Huang et al., 2013; Li et al., 2015). In sepsis, several studies have described the lncRNA profile in inflammatory injured cells in vitro (Ilott et al., 2014; Lin et al., 2015; Singh et al., 2016), but the lncRNA landscape in sepsis patients remains unexplored.
In this study, we investigated lncRNA expression profiles in the peripheral blood of sepsis patients and healthy volunteers, allowing us to screen for potential biomarkers of sepsis.
Materials and Methods
Participants and ethical approval
In this study, 22 consecutive ICU patients newly diagnosed with sepsis (Table 1) and 22 healthy volunteers from the Chinese PLA General Hospital were recruited from February 2014 to May 2014. Sepsis was diagnosed based on the 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions (Levy et al., 2003). Exclusion criteria were as follows: (1) presence of tumors; (2) age less than 16 years; (3) infection with AIDS or HIV; and (4) rejection of participation in this study. Blood samples were collected within 24 h of sepsis diagnosis. The Ethics Committee of Chinese PLA General Hospital approved this study. Written informed consent was obtained from all participants or their families.
1 = dead; 0 = survive.
APACHE II, acute physiology and chronic health evaluation II; CRP, C-reactive protein; Hb, hemoglobin; PCT, procalcitonin; SOFA, sequential organ failure assessment; WBC, white blood cell.
RNA isolation and quality assessment
Total RNA from peripheral blood was isolated and purified with a TRIpure LS Reagent Kit according to the manufacturer's instructions (BioTeke, Beijing, China). The quantity and quality of RNA were measured by absorbance at 260 nm using a spectrophotometer (NanoDrop). RNA integrity was assessed by agarose gel electrophoresis.
Microarray and computational analysis
Total RNA from three patients (A01-03) and three volunteers (B01-03) underwent lncRNA microarray screening. CapitalBio Corporation provided human lncRNA microarray service using the lncRNA+mRNA Human Gene Expression Microarray V3.0 (Beijing, China). This array contained ∼37,000 lncRNA probes and 34,000 mRNA probes. All lncRNA sequences were collected from 13 authoritative databases, including GENCODE, Ensembl, RefSeq, UCSC, H-InvDB, and Human lincRNA catalog. Agilent Feature Extraction software was used to analyze and extract data from the acquired array images (v10.7; Agilent Technologies). The extracted data were quantile-normalized and analyzed using GeneSpring software (Agilent Technologies). Genes with Benjamini-Hochberg false discovery rate (FDR) corrected p-values of 0.05 and threshold values of ≥2- and ≤2-fold change were considered to be differentially expressed.
Real-time quantitative polymerase chain reaction
Total RNA was retrotranscribed to cDNA using a PrimeScript™ RT Reagent Kit with a gDNA Eraser according to the manufacturer's instructions (TaKaRa, Japan).
A 20 μL volume reaction was used in real-time quantitative polymerase chain reaction (qRT-PCR) containing 1 μL cDNA, 1 μL 10 μM primer, 10 μL 2× KAPA SYBR FAST qRT-PCR Master Mix Universal, and 8 μL H2O (KAPA Biosystems, USA). Primers were designed using Primer BLAST (Table 2). Reaction settings consisted of a predenaturation step for 3 min at 95°C followed by 40 cycles of denaturation and primer annealing and extension for 3 s at 95°C and 30 s at 60°C. Samples were heated for 1 min at 95°C and 1 min at 60°C and then heated from 55°C to 100°C for melt curve. Following amplification, cycle thresholds (Ct) were obtained to calculate fold differences in gene expression using the 2−ΔΔCt method.
lncRNA, long noncoding RNA.
Coexpression analysis and functional enrichment analysis
To evaluate coexpression mRNAs of selected lncRNA, Pearson correlation coefficients (PCCs) were calculated. PCCs between lncRNA and mRNAs with no less than 0.99 were considered significant correlation pairs. Based on those significant correlation mRNAs, functional enrichment analyses were conducted using KOBAS 2.0 (KEGG Orthology Based Annotation System) (Xie et al., 2011).
Statistical analyses
Data are expressed as mean ± standard deviation and were subjected to a Mann-Whitney test. Statistical analyses were performed using SPSS 19.0 software (IBM) and Prism 5 (GraphPad Software). A difference of p < 0.05 was considered to be statistically significant.
Results
Overview of differentially expressed lncRNAs in sepsis
We evaluated lncRNA expression of three sepsis patients (A01-03) and three healthy volunteers (B01-03) using lncRNA microarray. The hierarchical clustering indicated a significant difference in lncRNA profile between the two groups (Fig. 1A). Among 1316 differentially expressed lncRNAs (≥2-fold change, p < 0.05), 771 were downregulated and 545 were upregulated in the sepsis group compared to the healthy group (Fig. 1B); 25 differed more than 20-fold (Table 3).
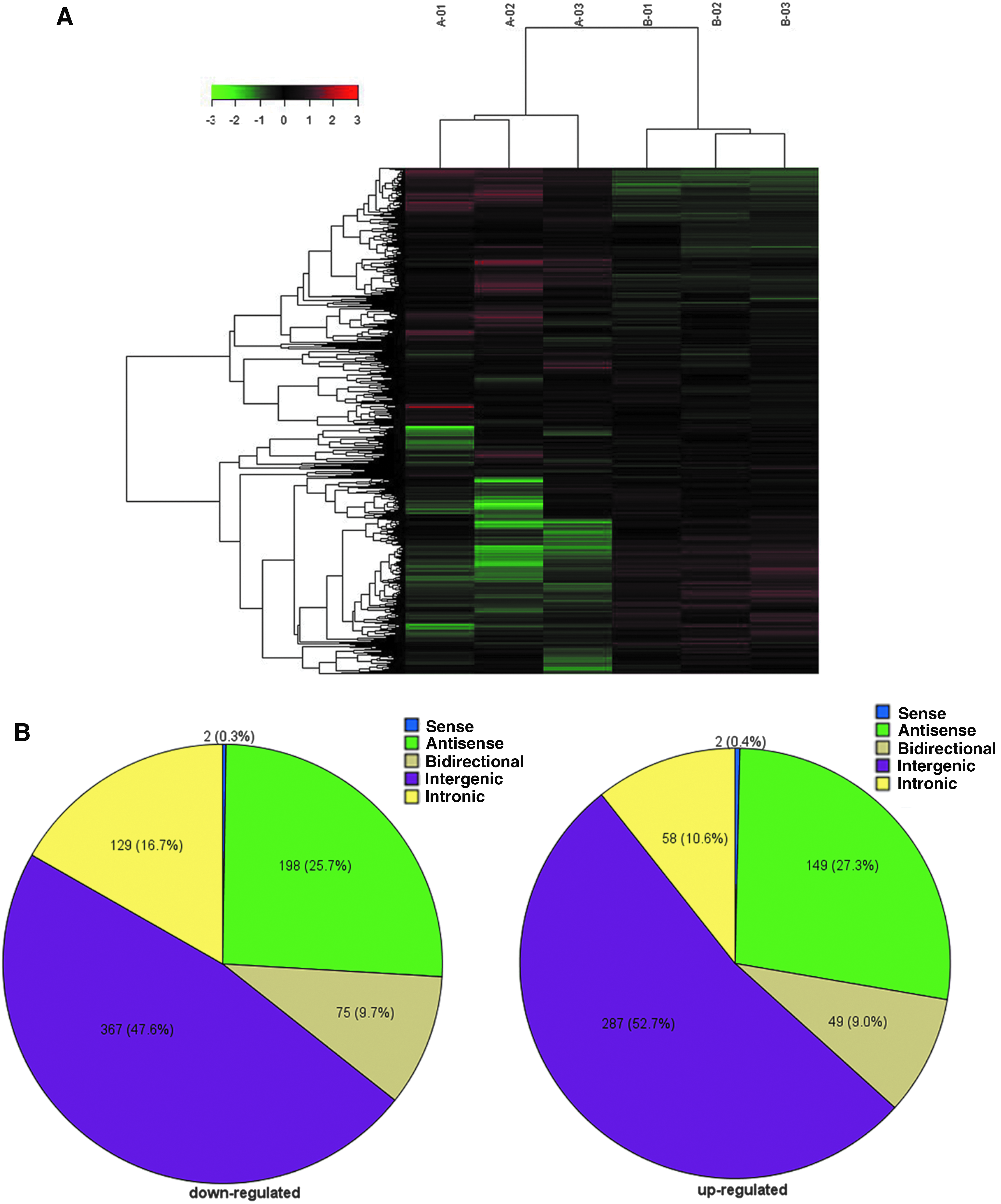
lncRNA expression profile in sepsis.
lncRNA confirmation and selection by qRT-PCR
Based on fold change and p-value, we selected ENST00000504301.1, ENST00000452391.1, uc001vji.1, and uc021zxw.1 for further validation. To confirm the reliability of the microarray results, expression of those four lncRNAs was retested using qRT-PCR in the same three sepsis samples (A01-03) and three healthy samples (B01-03). lncRNA expression results from qRT-PCR were in accord with microarray results (Fig. 2A). The fold changes using qRT-PCR were 513 for ENST00000504301.1, 7.58 for ENST00000452391.1, 5.93 for uc001vji.1, and 77 for uc021zxw.1.

Expression levels of uc001vji.1, uc021zxw.1, ENST00000452391.1, and ENST00000504301.1 using qRT-PCR.
We then investigated the expression of those four lncRNA in all 22 sepsis patients and 22 healthy volunteers. When samples were enlarged, the expression of lncRNA uc001vji.1 (p < 0.001), uc021zxw.1 (p < 0.001), and ENST00000452391.1 (p < 0.001) still differed significantly, whereas that of lncRNA ENST00000504301.1 (p = 0.213) was not statistically significant (Fig. 2B). Receiver operating characteristic (ROC) curve analyses of lncRNA uc001vji.1, uc021zxw.1, and ENST00000452391.1 were further performed to evaluate their diagnostic values in sepsis (Fig. 2C). Area under ROC curve (AUC) of uc001vji.1, uc021zxw.1, and ENST00000452391.1 was as follows: 0.971 (95% confidence interval [CI]: 0.921-1.000), 0.961 (95% CI: 0.907-1.000), and 0.866 (95% CI: 0.761-0.970) separately. These results indicated that those three lncRNAs were potential biomarkers of sepsis.
Potential prognostic relevance of selected lncRNAs
Among 22 sepsis patients, 16 survived and 6 did not survive. To evaluate potential prognostic relevance, expression levels of uc001vji.1, uc021zxw.1, ENST00000504301.1, and ENST00000452391.1 between survivor and nonsurvivor groups were analyzed. As shown in Figure 3, ENST00000504301.1 had a decreased expression (p = 0.022) and ENST00000452391.1 had an increased expression (p = 0.033) in nonsurvivor group than survivor group, whereas uc001vji.1 (p = 0.883) and uc021zxw.1 (p = 0.185) were not differentially expressed.
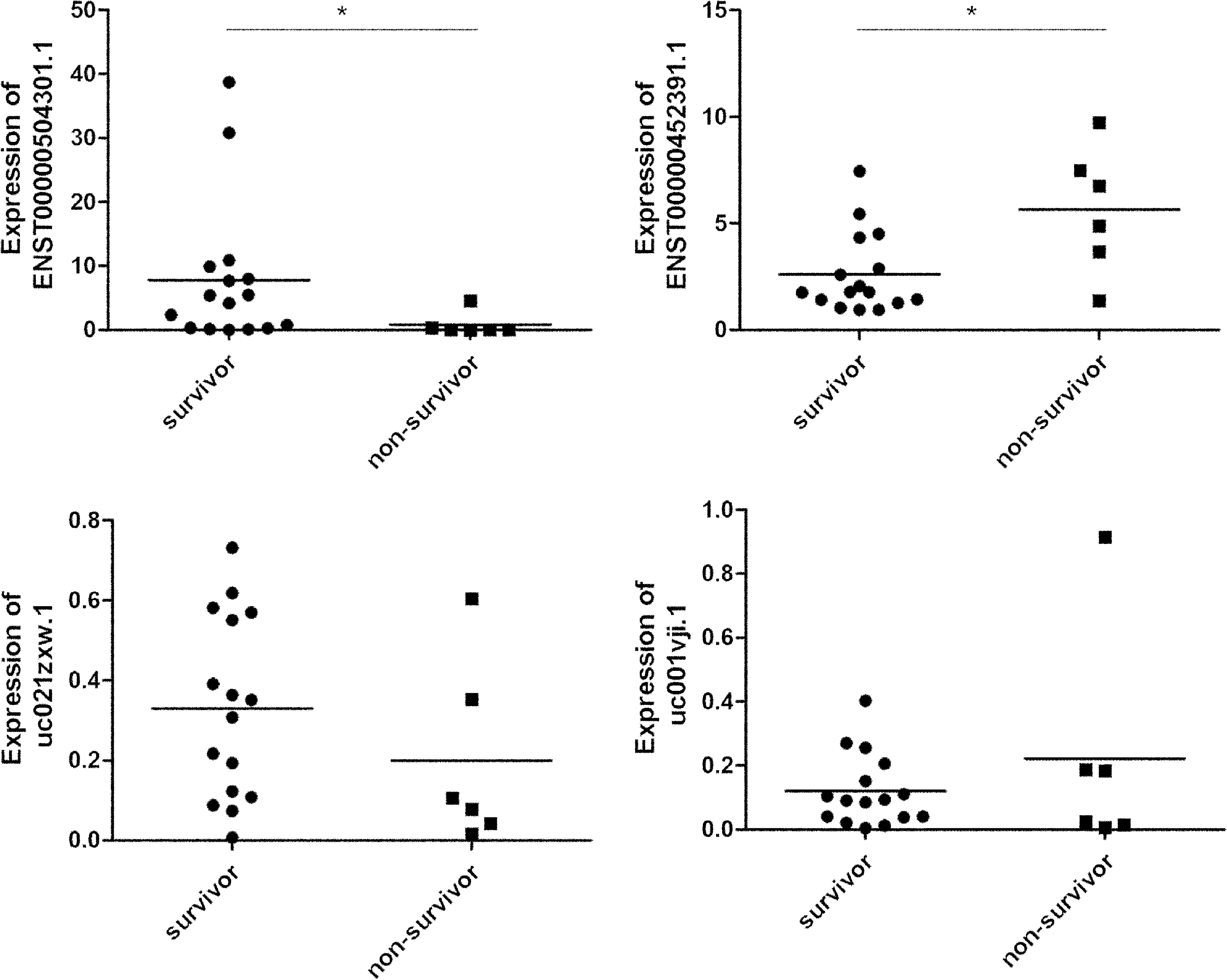
Potential prognostic value of ENST00000452391.1, ENST00000504301.1, uc001vji.1, and uc021zxw.1. *p < 0.05; survivor group (n = 16) vs. nonsurvivor group (n = 6).
Functional enrichment analysis of ENST00000452391.1
Since ENST00000452391.1 had both potential diagnostic and prognostic value, its coexpression mRNAs and bioinformatics analysis were performed. One hundred seventy-nine mRNAs were selected in the coexpression network with the criterion that PCCs were higher than 0.99 or less than −0.99 (Fig. 4A).

Functional analysis of ENST00000452391.1
Based on the coexpression mRNAs, gene ontology (GO) and Pathway analysis of ENST00000452391.1 were conducted. The top 10 most significant down- or upregulated enriched GO terms are shown in Figure 4B. The top 10 most significant down- or upregulated enriched pathway terms are shown in Figure 4C.
Discussion
lncRNAs are emerging as key players in various biological processes involved in numerous human diseases (Sun and Kraus, 2015; Schmitz et al., 2016). Although sepsis-related lncRNAs have been less well studied, several lncRNAs have been found to be inflammatory regulators. For example, LincRNA-Cox2 can mediate both the activation and repression of innate immune genes and take part in inflammatory response regulation (Carpenter et al., 2013). Lethe can inhibit NF-κB-dependent inflammatory signaling in a dose-dependent manner (Rapicavoli et al., 2013), and THRIL, NEAT1, lnc-IL7R, and NeST contribute to the inflammatory response by regulating expression of anti-inflammatory cytokines such as TNF-α, VCAM-1, IL-6, IL-8, and IFN-γ (Gomez et al., 2013; Cui et al., 2014; Imamura et al., 2014; Li et al., 2014). In addition, differentially expressed lncRNA profiles in vitro were found in lipopolysaccharide (LPS)-induced human umbilical vein endothelial cells and human monocytes (Ilott et al., 2014; Singh et al., 2016). Both studies suggest that LPS can induce comprehensive changes in lncRNA expression. Although those studies have not mentioned sepsis, those lncRNAs may take part in sepsis process since sepsis is a systemic inflammatory response.
To our knowledge, this study demonstrated the lncRNA expression profile in the peripheral blood of sepsis patients for the very first time. Sepsis patients and healthy volunteers were well clustered based on 1316 significantly differentially expressed lncRNAs (771 downregulated and 545 upregulated). To validate the microarray results, qRT-PCR of uc001vji.1, uc021zxw.1, ENST00000504301.1, and ENST00000452391.1 was performed. These qRT-PCR data matched well with the microarray results. According to the relative position to the neighboring protein-coding transcript, lncRNAs can be divided into five broad categories as follows: sense, antisense, bidirectional, intronic, and intergenic (Ponting et al., 2009). Approximately half of differentially expressed lncRNAs in sepsis patients belonged to the intergenic subcategory, which is located within the genomic interval between two genes and could control gene expression through interaction with various chromatin-modifying complexes (Ulitsky and Bartel, 2013; Deniz and Erman, 2017).
An ideal biomarker of sepsis is still yet to be uncovered. Among the more than 170 previously proposed sepsis biomarkers, C-reactive protein and procalcitonin are the most widely used in the clinic (Pierrakos and Vincent, 2010). However, their poor specificity makes it difficult to distinguish sepsis from other inflammatory conditions (Clyne and Olshaker, 1999; Simon et al., 2004; Tang et al., 2007). With the development of genomic approaches, circulating noncoding RNAs, including microRNAs, lncRNAs, and circular RNAs (circRNAs), are becoming promising components of biomarkers. In the field of sepsis, previous studies have focused on microRNAs, while no study has evaluated the possibility of using lncRNAs or circRNAs as biomarkers (Dumache et al., 2015; Benz et al., 2016; Ho et al., 2016).
In this study, we selected uc001vji.1, uc021zxw.1, ENST00000504301.1, and ENST00000452391.1 as biomarker candidates. Since only three pair samples were tested using microarray methods, the microarray results may not represent the sepsis population well. qRT-PCR was therefore also performed in 22 expended pair samples and showed that uc001vji.1, uc021zxw.1, and ENST00000452391.1 had significant differential expressions, while ENST00000504301.1, the most (∼64-fold) dysregulated lncRNA in microarray result, was not different (p > 0.05) between sepsis patients and healthy volunteers. With more than 0.8 AUC values, uc001vji.1, uc021zxw.1, and ENST00000452391.1 may be favorable candidates for diagnostic biomarkers in sepsis. Therefore, we then stratified the sepsis population on the basis of patient outcome. Survivors expressed a higher level of ENST00000504301.1 and a lower level of ENST00000452391.1 than nonsurvivors, indicating that these two lncRNAs are connected with sepsis outcome. Considering the small sample size of each subgroup, these connections must be further verified.
ENST00000452391.1 was the only lncRNA associated with both sepsis diagnosis and prognosis, suggesting that it may play a critical role in sepsis. Functional enrichment analysis based on its coexpression mRNAs was hence performed. GO terms which ENST00000452391.1 mainly involved, such as pathogenesis, lymphocyte activation, leukocyte cell-cell adhesion and leukocyte activation, are also crucial procedures in sepsis development and progression. We will confirm the association between ENST00000452391.1 and sepsis and further study its mechanism in our future work.
In conclusion, we revealed for the first time the lncRNA expression profile in sepsis patients and identified four circulating lncRNAs as potential candidates of sepsis biomarkers. ENST00000452391.1 may play a crucial role in the development and progression of sepsis. Further study is needed to verify the connection and to explore the mechanism.
Footnotes
Acknowledgments
The Key Projects in the Chinese PLA Science & Technology Program during the Twelfth Five-year Plan Period (No. BWS11J057) supported this study.
Author Disclosure Statement
No competing financial interests exist.