
Abstract
Background:
Noncoding RNAs (ncRNAs), primarily microRNAs and long ncRNAs, play important roles in lung cancer. However, the role of long ncRNA (lncRNA)-X-inactive specific transcript (XIST) in non-small-cell lung cancer (NSCLC) is unclear. The purpose of this study was to explore the biologic function and potential mechanism of XIST in NSCLC progression.
Materials and Methods:
XIST, miR-137, and Notch-1 expression were detected by quantitative real-time PCR (qRT-PCR). Levels of proliferation- and epithelial-mesenchymal transition (EMT)-related proteins were assessed by Western blot. The correlations between XIST and miR-137, as well as miR-137 and Notch-1, were evaluated by bioinformatic analysis and luciferase reporter assays.
Results:
We confirmed that XIST is aberrantly upregulated in NSCLC tissues and cell lines. XIST depletion inhibited cell proliferation and TGF-β1-induced EMT in A549 and H1299 cells. Spearman's correlation analysis showed an inverse correlation between miR-137 and XIST in NSCLC tissues, and miR-137 levels were found to be aberrantly reduced in A549 and H1299 cells. Furthermore, XIST could act as an endogenous sponge by directly binding to miR-137, negatively regulating its expression. miR-137 overexpression inhibited proliferation and TGF-β1-induced EMT in A549 and H1299 cells, whereas XIST could reverse the inhibitory effect of miR-137 on proliferation and TGF-β1-induced EMT. In addition, Notch-1 was identified as a direct target gene of miR-137, with the XIST-miR-137 axis regulating activation of the Notch-1 pathway.
Conclusion:
We identified a branch of the XIST/miR-137/Notch-1 pathway that regulates proliferation and TGF-β1-induced EMT in NSCLC, which could be involved in NSCLC progression.
Introduction
N
Metastasis remains the principal cause of death in lung cancer patients and epithelial-mesenchymal transition (EMT) is well known to be an initiating and vital event in the metastatic cascade, in which epithelial cancer cells lose polarity and cell-cell contacts, and acquire mesenchymal features that promote migration and invasion (van Denderen and Thompson, 2013). TGF-β signaling is a main inducer of EMT in lung cancer (Liu et al., 2014a).
Long noncoding RNAs (lncRNAs) are a class of noncoding RNAs (ncRNAs), with more than 200 bases, which play vital roles in many cancers, including NSCLC (Tantai et al., 2015). Recently, it was reported that lncRNAs, including lncRNA-X-inactive specific transcript (XIST), LINC00473, NEAT1, AGAP2-AS1, TATDN1, and LINC01133, are involved in NSCLC progression (Fang et al., 2016). The XIST transcript is a product of the XIST gene, which is highly expressed, and is an oncogenic lncRNA that drives tumorigenesis in NSCLC (Fang et al., 2016). However, the precise molecular mechanisms of XIST in NSCLC are still unclear.
MicroRNAs (miRNAs) are 19- to 25-nucleotide regulatory ncRNAs that bind to partially complementary recognition sequences of mRNA, causing either degradation or inhibition of translation, thus effectively repressing target gene expression at the posttransciptional level (Vennin et al., 2015). miRNAs have been implicated in tumorigenesis, metastatic progression, and therapeutic responses (Garofalo and Croce, 2013; Di Leva et al., 2014). It has been reported that miR-137 is downregulated in human lung cancer tissues and acts as a tumor suppressor (Shen et al., 2016). However, the molecular mechanism of miR-137 suppression in NSCLC remains unknown.
Growing evidence indicates that ncRNAs, especially lncRNAs and miRNAs, play vital roles in tumorigenesis, partially by regulating EMT (Chen et al., 2017b). Recently, it has been reported that XIST acts as a competing endogenous RNA (ceRNA), which negatively regulates the target gene (miR-137) by directly sponging miR-137, thus contributing to glioma tumorigenesis (Wang et al., 2017). Hence, we focused on the roles and underlying mechanisms of XIST and miR-137 in the ceRNA regulatory network of NSCLC.
Materials and Methods
Patients and specimens
Thirty-three NSCLC tissue and corresponding adjacent nontumor tissue samples were obtained from patients diagnosed with NSCLC by pathologists from the First Affiliated Hospital of Zhengzhou University. Of these 33 samples, 21 patients were diagnosed with adenocarcinoma NSCLC, 4 patients had squamous cell carcinomas, and 13 patients had other types of cancer. Patients were male with a mean age at lung cancer diagnosis of 61 years; 85% of patients reported a history of tobacco smoking and 35% of patients smoked at the time of diagnosis. Eight patients had stage I disease, 3 patients had stage II disease, 11 patients had stage III disease, and 10 had stage IV disease based on histology at diagnosis.
All samples were collected from patients who had not received treatment before surgery. Samples were immediately snap-frozen in liquid nitrogen and stored at −80°C for further analysis. The experiment protocols were performed with the approval of Ethics Committee of the First Affiliated Hospital of Zhengzhou University, and informed consent was obtained from the patients.
Cell culture and transfection
Human lung cancer cell lines (A549, H358, H460, H1299, and PC9) and normal bronchial epithelial cell line 16HBE were purchased from the American Type Culture Collection (ATCC; Manassas, VA). All human lung cancer cells were grown in RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA), supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific), penicillin, and streptomycin (Thermo Fisher Scientific). 293T cells (ATCC) were grown in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with 10% FBS and 1% penicillin-streptomycin solution. All cells were incubated in a humid atmosphere containing 5% CO2 at 37°C.
TGF-β1 was purchased from R&D system (Minneapolis, MN). Cells were treated with TGF-β1 (2 ng/mL) for the indicated time. miR-NC, miR-137 mimics (miR-137), pcDNA-XIST, and pcDNA empty vector were purchased from Shanghai GenePharma Company (Shanghai, China), and were transfected into cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Lentivirus production and infection
The pLVX vector containing shRNA XIST or the control sequence (GenePharma) was cotransfected with Lenti-Pac HIV Expression Packaging Mix (GenePharma) into 293T cells using Lipofectamine 2000. Lentiviral particles in the supernatant 48 h posttransfection were harvested and then filtered by centrifugation at 500 g for 10 min.
The A549 and H1299 cells were then infected with LV-sh-XIST or LV-sh-NC. The cells were treated with puromycin (2 μg/mL) for 2 weeks to gain the stably transfected cells that were picked as sh-XIST and sh-NC and then used for subsequent assays.
RNA isolation and quantitative real-time PCR
Total RNA from frozen tissue samples and cells was extracted using TRIzol reagent (Invitrogen) and miRNAs were isolated using a miRNA extraction kit (Ambion, Carlsbad, CA), according to the respective manufacturer's instructions. The concentration and purity of total RNA were detected using the NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE). Quality of the isolated RNA was confirmed to be suitable for reverse transcription into cDNAs by assessing the 28S/18S rRNA ratio and RNA integrity number. The 28S/18S rRNA ratio of RNA was ∼2.0 and the RNA integrity number was >7, which is considered to be high quality.
Total RNA and lncRNA were reverse transcribed into cDNAs using Prime Script TM RT Master Mix (TaKaRa Bio Technology, Dalian, China). The cDNA template was amplified by real-time reverse transcription-polymerase chain reaction (RT-PCR) using the SYBR Premix Dimmer Eraser kit (TaKaRa). The reverse transcription for miRNA was performed using One Step PrimeScript® miRNA cDNA Synthesis Kit (TAKARA) and quantitative RT-PCR (qRT-PCR) analysis was performed with the SYBR Premix Ex Taq II (TAKARA); U6 and β-actin were used as controls. mRNA levels were determined using the relative quantification 2−ΔΔCT method.
Western blot
Total protein from tissues and cultured cells was lysed in RIPA buffer (Sigma-Aldrich) and was quantified using a BCA protein assay kit (Beyotime Institute of Biotechnology, Haimen, China). Then, proteins were run on a 10% SDS-PAGE and transferred to PVDF membranes (Millipore, Bedford, MA). The membranes were blocked with 5% nonfat milk in TBST for 1 h at room temperature and incubated with primary antibodies at 4°C overnight. All membranes then were probed with the secondary antibody for 2 h at room temperature.
Antibodies used in this study were as follows: Ki67 (Dako, Glostrup, Denmark), E-cadherin (Cell Signaling Technology, Danvers, MA), N-cadherin (Cell Signaling Technology), vimentin (Cell Signaling Technology), Notch-1 (Cell Signaling Technology), Cyclin D1 (Cell Signaling Technology), Survivin (Cell Signaling Technology), β-actin (Cell Signaling Technology), and HRP-labeled secondary antibody (Santa Cruz). Signals were assessed and visualized by the ECL system (Amersham, Piscataway, NJ). The integrated density of proteins was quantified using Image J software (NIH).
MTT assays
A549 and H1299 cells were seeded into 96-well culture plates (Costar, Corning, NY) at a density of 3 × 103 cells/well. At indicated times (0, 24, 48, and 72 h) posttransfection, 20 μL of 5 mg/mL 3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT; Sigma, St. Louis, MO) was added to each well and the cells were incubated for another 4 h at 37°C. After 4 h of incubation, the supernatant in each well was removed and 100 μL of dimethyl sulfoxide (DMSO; GIBCO, New York) was added to solubilize the formazan salt. The absorbance at 490 nm was measured by using a UV microplate reader (Tecan Austria GmbH, Groedig, Austria).
Luciferase reporter assays
Mutations of the miR-137 binding sites within the XIST luciferase reporter vector (mutated XIST [XIST-MUT]) were introduced by site-directed mutagenesis and the resulting plasmid was referred to as XIST-MUT. Mutations of the miR-137 binding sites within the Notch-1 luciferase reporter vector (mutated Notch-1) were introduced by site-directed mutagenesis and the resulting plasmid was referred to as Notch-1-MUT. A549 and H1299 cells were seeded into a 24-well plate 24 h before cotransfection with miR-137 or miR-NC and XIST-WT (wild-type XIST) or XIST-MUT, miR-137 or miR-NC and Notch-1-WT (wild-type Notch-1) or Notch-1-MUT (mutated Notch-1), or miR-137 and pcDNA or pcDNA-XIST and Notch-1-WT, or Notch-1-MUT. At 48 h posttransfection, luciferase reporter assays were performed using the Dual Luciferase Reporter Assay System (Promega, WI) according to the manufacturer's protocol.
Statistical analysis
All experiments were repeated at least thrice independently. All data are shown as mean ± standard deviation. Student's t-test and one-way ANOVA were employed to determine significance, and p < 0.05 was considered statistically significant.
Results
XIST was upregulated in NSCLC tissues and cell lines
XIST levels in 33 paired NSCLC tissue and adjacent healthy tissue samples were detected by qRT-PCR. As shown in Figure 1A, XIST levels were significantly increased in NSCLC tissues in comparison with the control group. XIST expression was also significantly increased in NSCLC cell lines (A549, H358, H460, H1299, and PC9) compared with normal bronchial epithelial cell line 16HBE (Fig. 1B). Among these cells, A549 and H1299 cells showed the highest level of XIST and thus were chosen for subsequent analyses. These results implied that XIST might be involved in NSCLC progression.

XIST was significantly upregulated in NSCLC tissues and cell lines.
Knockdown of XIST suppressed cell proliferation and TGF-β1-induced EMT in A549 and H1299 cells
To explore the biological roles of XIST in NSCLC progression, A549 and H1299 cells were first transfected with sh-XIST or sh-NC (Fig. 2A). MTT assay revealed that XIST knockdown significantly inhibited cell proliferation of A549 (Fig. 2B) and H1299 (Fig. 2C) cells at 48 and 72 h compared with the control group. Moreover, Western blot analysis revealed that Ki67 levels were significantly reduced in sh-XIST-transfected A549 and H1299 cells compared with the sh-NC group (Fig. 2D).
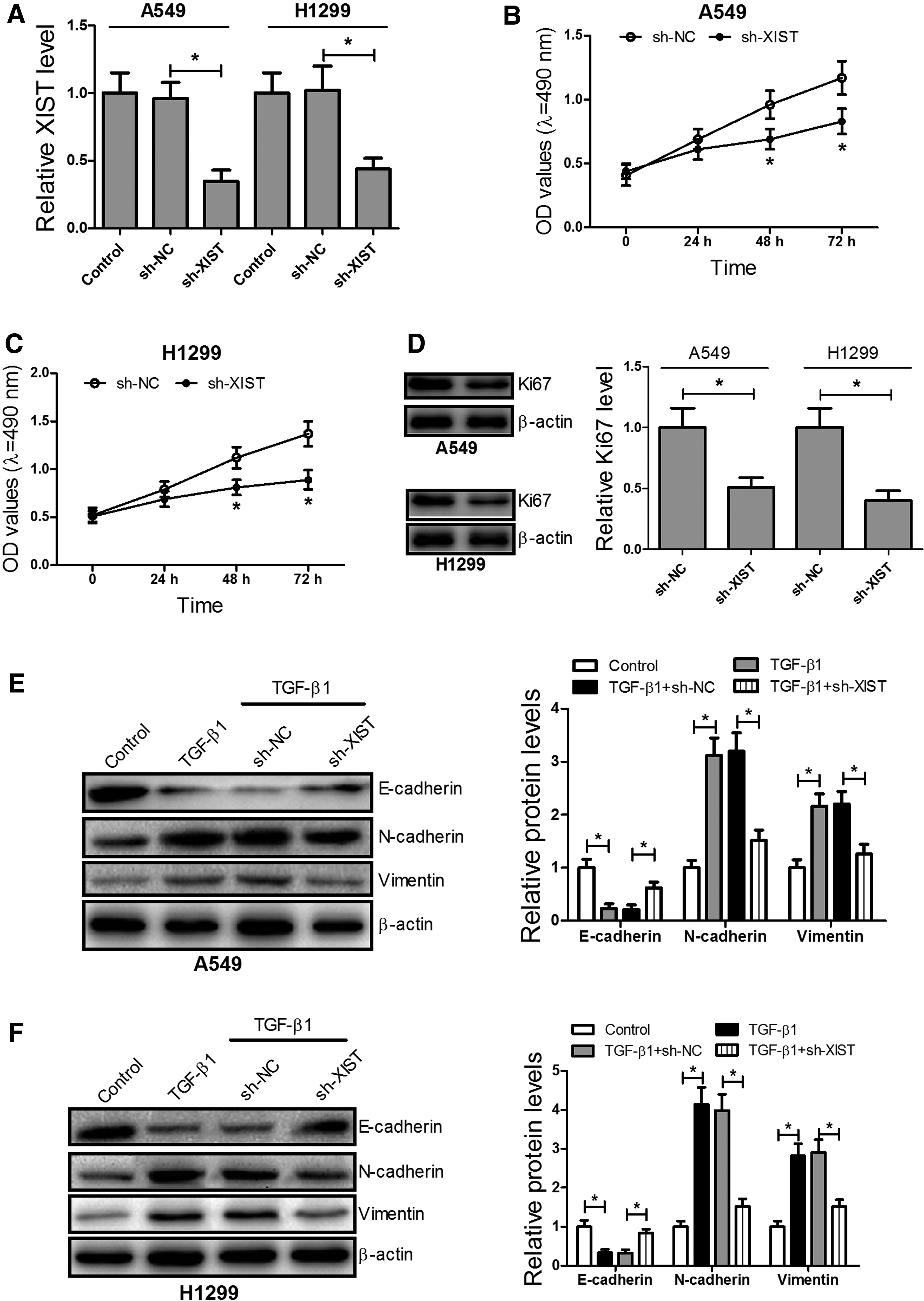
Knockdown of XIST inhibited proliferation and TGF-β1-induced EMT in A549 and H1299 cells.
It has been reported that EMT plays a key role in cell migration and invasion of cancer cells (Chen et al., 2017a). Western blot analysis showed that XIST knockdown significantly overturned TGF-β1-induced loss of E-cadherin and production of N-cadherin, as well as vimentin in A549 and H1299 cells (Fig. 2E, F). These results suggest that knockdown of XIST can inhibit cell proliferation and TGF-β1-induced EMT in A549 and H1299 cells.
XIST directly interacted with miR-137 and negatively regulated its expression in A549 and H1299 cells
A recent study reported that XIST can bind to miR-137 and affect miR-137 function in glioma (Yu et al., 2017). As shown in Figure 3A, we found a significant negative correlation between XIST and miR-137 expression, which revealed that miR-137 expression was significantly reduced in A549 and H1299 cells compared with the control group (Fig. 3B). In addition, bioinformatics analysis using the starBase v2.0 (http://starbase.sysu.edu.cn/) and Miranda (www.microrna.org/) predicted that there existed a binding site of miR-137 in XIST (Fig. 3C).
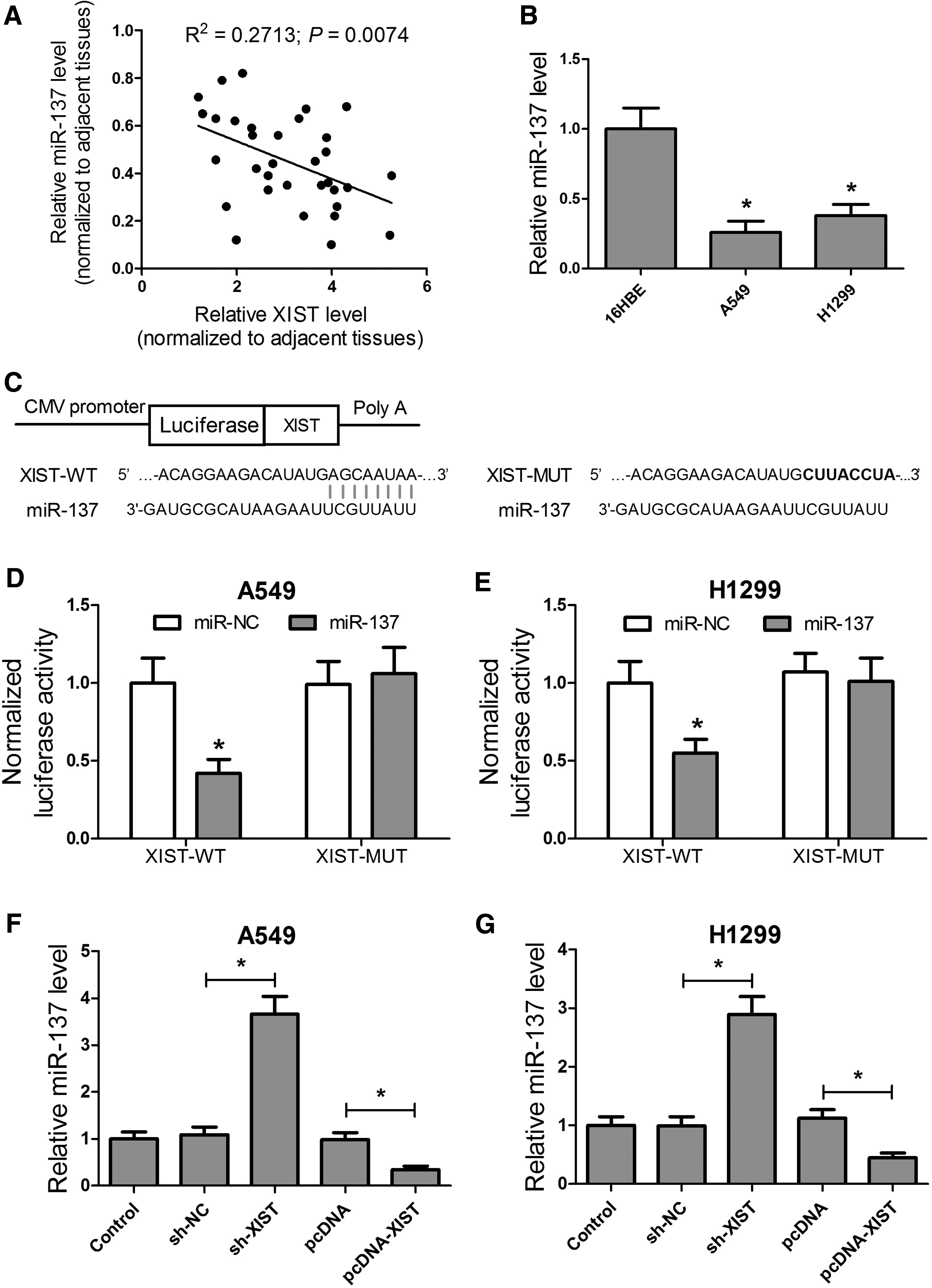
XIST directly interacted with miR-137, which negatively regulates its expression in A549 and H1299 cells.
To determine whether XIST was bound to miR-137 in A549 and H1299 cells, dual luciferase reporter assays were performed, which showed that miR-137 could directly bind the XIST sequence (Fig. 3D, E). To further confirm the luciferase assay results, qRT-PCR analysis revealed that XIST knockdown significantly upregulated miR-137 levels, while XIST overexpression significantly inhibited miR-137 expression in both A549 and H1299 cells (Fig. 3F, G). These data indicate that XIST directly interacted with miR-137 and negatively regulated its expression in A549 and H1299 cells.
XIST-miR-137 axis regulated cell proliferation and TGF-β1-induced EMT in A549 and H1299 cells
To further investigate whether XIST affected cell proliferation and TGF-β1-induced EMT in A549 and H1299 cells by sponging to miR-137, MTT assay and Western blot assays were performed. The MTT assay showed that miR-137 overexpression significantly restrained cell proliferation of A549 (Fig. 4A) and H1299 (Fig. 4B) cells at 48 and 72 h compared with the control group, whereas XIST overexpression reversed the inhibitory effect of miR-137 on cell proliferation of A549 and H1299 cells. Western blot analysis revealed that Ki67 expression was significantly suppressed in miR-137-transfected A549 and H1299 cells compared with the miR-NC group, while XIST overexpression weakened the inhibitory effect of miR-137 on Ki67 expression (Fig. 4C, D). Western blot analysis also showed that miR-137 overexpression significantly alleviated the loss of E-cadherin and production of N-cadherin, as well as vimentinis in A549 cells induced by TGF-β1 and H1299 cells, while XIST overexpression reversed the effects of miR-137 overexpression on TGF-β1-induced reduction of E-cadherin and increased N-cadherin, as well as vimentinis (Fig. 4E, F). These results suggest that the XIST-miR-137 axis regulates cell proliferation and TGF-β1-induced EMT in A549 and H1299 cells.

The XIST-miR-137 axis modulated proliferation and TGF-β1-induced EMT in A549 and H1299 cells. MTT assay was performed to detect cell proliferation of A549
The XIST-miR-137 axis regulated activation of the Notch signaling pathway in A549 and H1299 cells
TargetScan and miRBase software were used to predict the target gene of miR-137, which identified Notch-1 as a potential target gene (Fig. 5A). To determine whether miR-137 directly binds to Notch-1 in A549 and H1299 cells, dual luciferase reporter assays were performed. These results showed that miR-137 could directly bind Notch-1 in A549 and H1299 cells (Fig. 5B, C). In addition, XIST knockdown significantly downregulated Notch-1 mRNA levels and XIST upregulation significantly increased Notch-1 mRNA levels in A549 cells (Fig. 5D). Moreover, miR-137 overexpression also significantly reduced mRNA levels of Notch-1, while XIST overexpression significantly reversed the inhibitory effect of miR-137 on Notch-1 mRNA expression in A549 cells.
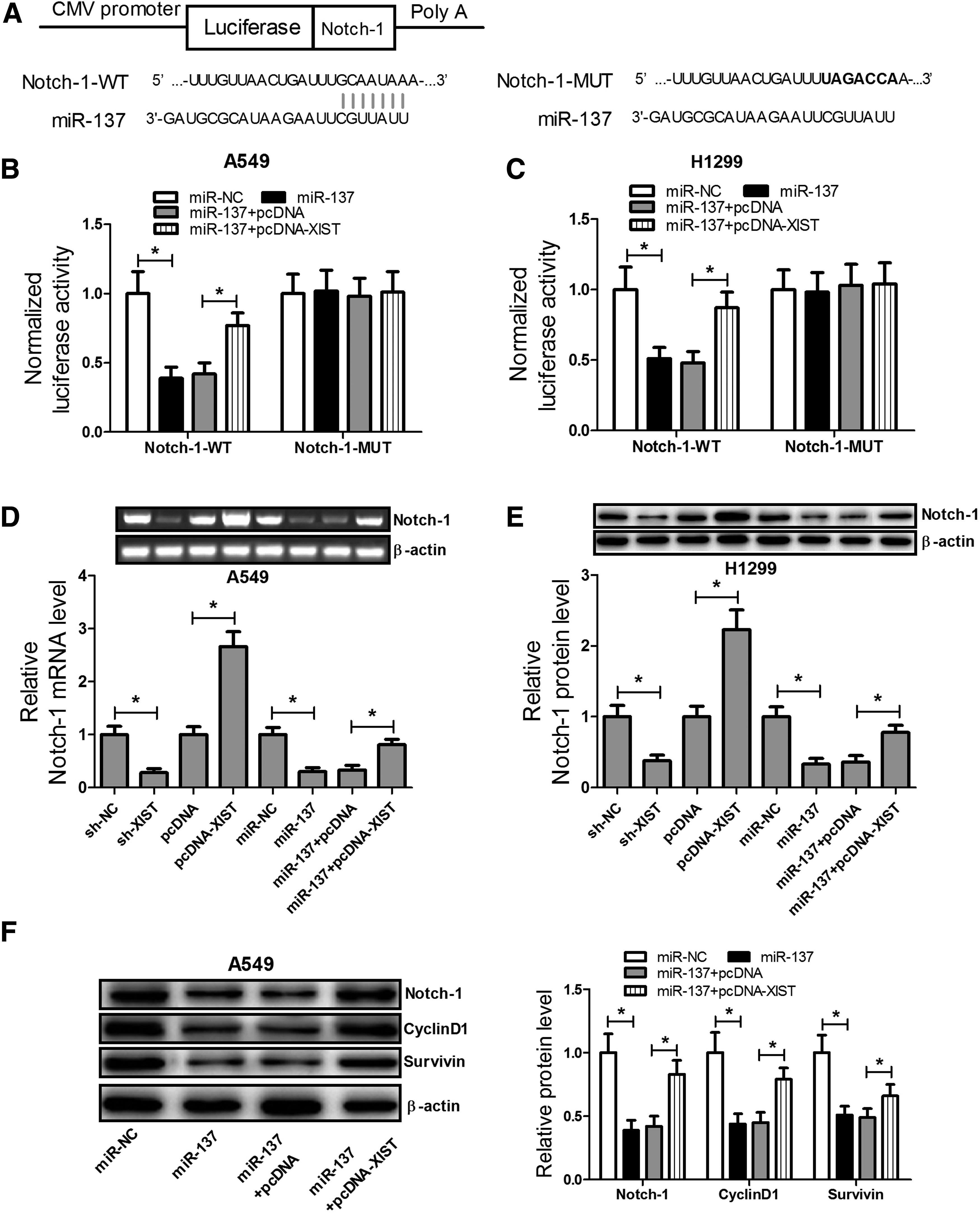
The XIST-miR-137 axis regulated the activation of the Notch signaling pathway in A549 and H1299 cells.
Western blot analysis revealed that XIST knockdown significantly inhibited Notch-1 protein expression and XIST upregulation significantly increased Notch-1 protein expression in A549 cells. miR-137 overexpression also significantly reduced protein levels of Notch-1, whereas XIST overexpression significantly reversed the inhibitory effect of miR-137 on Notch-1 expression in H1299 cells (Fig. 5E). In addition, Western blot analysis showed that miR-137 overexpression significantly suppressed the expression of Notch-1, Cyclin D1, and Survivin, whereas XIST overexpression significantly reversed the inhibitory effect of miR-137 on Notch-1, Cyclin D1, and Survivin expression (Fig. 5F). These results indicate that the XIST-miR-137 axis modulates activation of the Notch signaling pathway in A549 and H1299 cells.
Discussion
Despite great progress in the early detection of NSCLC, nearly 50% patients have developing distant metastases at diagnosis (Chheang and Brown, 2013). Therefore, prevention of metastasis and investigation of the potential mechanisms of cancer cell metastasis are crucial to prevent cancer progression.
EMT is a crucial event that occurs during cancer metastasis (Kang and Massague, 2004), and studies have identified distinct signaling pathways that regulate EMT (De Craene and Berx, 2013). TGF-β reduces E-cadherin expression, increases vimentin expression, and increases migration and invasion in lung cancer cells (Liu et al., 2014a). We found that TGF-β significantly suppressed expression of E-cadherin and promoted the expression of N-cadherin and vimentin. Furthermore, our results revealed that XIST could sponge to miR-137 and negatively modulate its expression, as well as affect expression of TGF-β1-induced EMT-related proteins in A549 and H1299 cells, which could play a vital role in NSCLC progression.
Several studies have reported that miRNAs are involved in various parts of cancer biology, including tumorigenesis, tumor progression, and metastasis (Wan et al., 2013; Zhou et al., 2014). miR-137 functions as a tumor suppressor in several human cancers, including neuroblastoma, ovarian cancer, glioblastoma, and lung cancer (Althoff et al., 2013; Guo et al., 2013; Zhu et al., 2013; Tamim et al., 2014), and miR-137 overexpression significantly suppresses GIST cell proliferation and invasion (Liu et al., 2014b). In this study, we showed that miR-137 overexpression significantly inhibited proliferation and TGF-β-induced EMT in A549 and H1299 cells.
In recent years, studies have shown that lncRNAs play vital roles in tumor occurrence, invasion, and metastasis by modulating gene expression and signaling pathways (Li et al., 2015). Some lncRNAs are involved in NSCLC progression, such as LINC00473 (Chen et al., 2016), LINC01133 (Zang et al., 2016), NEAT1 (Sun et al., 2016), and XIST (Fang et al., 2016). It has also been reported that XIST functions as a ceRNA and knockdown of XIST exerts tumor-suppressive effects in glioblastoma by sponging miR-152 (Yao et al., 2015). In hepatocellular carcinoma, XIST can also directly sponge miR-92b and inhibit proliferation and metastasis by targeting miR-92b (Zhuang et al., 2016). Our findings revealed that XIST can directly sponge miR-137 and downregulate its expression and that, XIST overexpression could reverse the inhibitory effects of miR-137 upregulation on cell proliferation and TGF-β-induced EMT in A549 and H1299 cells.
The oncogenic potential of aberrant Notch activation has been observed in lung tumorigenesis (Chen et al., 2011). It has been showed that inhibition of Notch-1 signaling could suppress cellular growth and induce apoptosis in HCC cells (Kunnimalaiyaan et al., 2015). Cyclin D1 and Survivin are downstream targets of Notch-1 and collectively play roles in many cancers, including NSCLC (Chen et al., 2011; Naganuma et al., 2012; Zhang et al., 2013). Our study revealed that miR-137 could directly bind to Notch-1 and negatively regulate its expression in A549 and H1299 cells. We further determined that miR-137 overexpression significantly reduced the expression of Notch-1, cyclin D1, and Survivin, whereas XIST overexpression reversed these roles. Therefore, we conclude that the XIST-miR-137-Notch-1 axis may play an important role in NSCLC progression.
Conclusion
We found that XIST expression was increased and miR-137 levels were reduced in NSCLC tissues and cells. XIST knockdown inhibited proliferation and TGF-β-induced EMT through a novel regulatory network, the XIST-miR-137-Notch-1 signaling pathway. These data may provide a better understanding of NSCLC progression and facilitate drug development.
Footnotes
Author Disclosure Statement
No competing financial interests exist.