
Abstract
Aim:
To analyze the variants of the potential causative genes in five Chinese patients with primary distal renal tubular acidosis (dRTA) from five unrelated families, and to explore their possible genotype-phenotype correlations, so as to raise the awareness of the disease.
Methods:
Variants were identified by next generation sequencing. Clinical features and biochemical findings at the first presentation, as well as at follow-up visits were also investigated. One hundred unrelated healthy subjects were selected to evaluate each of the novel mutations found in this study.
Results:
A total of seven different mutations in the ATP6V0A4, ATP6V1B1, and SLC4A1 genes, the three main causative genes of dRTA, were detected in 4/5 patients. In patient I a novel heterozygous intronic mutation (c.639 + 1G>A) in the ATP6V0A4 gene was identified along with a heterozygous nonsense variant (c.580C>T, p.Arg194*). Two novel heterozygous missense mutations of the ATP6V1B1 gene (c.409C>T, p.Pro137Ser; c.904C>T, p.Arg302Trp) were identified in patient II. In patient III 2 novel heterozygous duplications (c.1504dupT, p.Tyr502Leufs*22; c.2351dupT, p.Phe785Ilefs*28) were found. Thus, these three patients all were compound heterozygotes leading to dRTA. These findings are consistent with the known autosomal recessive inheritance pattern of this disease. Furthermore, a de novo heterozygous missense mutation previously reported (c.1765C>A, p.Arg589Ser) in the SLC4A1 gene was observed in patient IV. No mutations in any of the known dRTA-related causative genes were found in the patient V.
Conclusions:
In the present study we identified 7 mutations, including 5 novel variants, in the three genes previously correlated with dRTA, enriching the human gene mutation database (HGMD). In addition, our lack of findings in these three genes for patient V suggests that other genes may contribute to dRTA in some cases.
Introduction
I
Anion exchanger member 1 (AE1), encoded by the SLC4A1 gene located on chromosome 17q21, is primarily responsible for chloride/bicarbonate exchange across the membrane of red cells or the alpha-intercalated cell of the renal collecting duct (Kollert-Jons et al., 1993; Tanner, 1997). The dRTA with disabled AE1 is mostly presented in a pattern of AD inheritance in Caucasians, whereas mutations associated with AR dRTA usually have been found in homozygous or compound heterozygous conditions or in Southeast Asian ovalocytosis families of Southeast Asian origin (Khositseth et al., 2012; Zhang et al., 2012). The AR dRTA is reported more often with the ATP6V1B1 and ATP6V0A4 gene mutations affecting the H+-ATPase (the production of ATP6V1B1) in the apical cell membrane (Miura et al., 2013; Gao et al., 2014). The ATP6V1B1 gene is expressed by intercalated cells of the renal collecting duct and endolymphatic sac epithelia, accounting for the sensorineural deafness (SNHL) (Escobar et al., 2016); the ATP6V0A4 gene encodes transmembrane a4 subunit, which may be involved in H+ translocation or transport and/or assembly of the H+-ATPase (Miura et al., 2013).
So far, a total of 100 dRTA-related gene mutations have been reported (Human Gene Mutation Database [HGMD] Professional 2017.4), including 47 in ATP6V0A4, 34 in ATP6V1B1, and 19 in SLC4A1. Moreover, a recent report by Enerbäck et al. (2017) identified that three dRTA patients with early-onset SNHL carried two novel homozygous missense mutations in FOXI1 gene, which were predicted to affect the highly conserved DNA-binding domain. Both variants disrupted cell function at the transcriptional regulatory level. They prevented the transcription factor FOXI1 from binding to regulatory DNA cis-elements of target gene promoters, leading to a severely reduced expression of membrane transport proteins (AE1 and H+-ATPase). This also indicated that FOXI1 had become a new pathogenic gene for dRTA (Enerbäck et al., 2017). However, primary dRTA has been rarely described in a Chinese population with only 10 novel related mutations detected so far (Shao et al., 2010; Li et al., 2012; Zhang et al., 2012; Gao et al., 2014; Xu and Yang, 2017), of which five ones were found by our previous research (Shao et al., 2010; Gao et al., 2014).
Herein, we performed mutation analysis of causative genes in five patients with primary dRTA from five unrelated Chinese families, and assessed the correlation between genotype and phenotype, expecting to further reveal the characteristics of mutations in Chinese patients with primary dRTA.
Materials and Methods
Patients
A total of five probands (two females and three males) with primary dRTA from five unrelated Chinese families who had been hospitalized in our nephrology department of the affiliated hospital of Qingdao University from 2014 to 2017 were recruited in this study. Their clinical features and representative biochemical data at the first admission are shown in Table 1. Necessary diagnostic criteria of primary dRTA included metabolic acidosis with a normal anion gap and inappropriate alkaline urine (pH >6.0) in a context of acidosis (CO2CP <18 mmol/L); normal fractional excretion rate of bicarbonate (
The 50th percentile of Chinese children normal reference values for height and weight of different ages and genders at diagnosis.
S, serum; U, urinary; N, normal.
The study protocol was approved by the Ethics Committee of the affiliated hospital of Qingdao University. Informed consent was obtained from each patient and his or her parents for the study and publication of material before participation.
Mutation analysis
DNA extraction
Peripheral blood DNA was extracted from the probands, their family members, and 100 normal healthy controls using GenElute blood genomic DNA Kit (Sigma, NA2010) according to manufacturer's instructions.
Next generation sequencing
A custom next generation sequencing (NGS) panel was used to analyze the exon regions and flanking intronic regions in more than 7000 genes from 5 probands. Generated reads were aligned to the human reference genome hg19 (University of California Santa Cruz [UCSC], http://genome.ucsc.edu) and National Center for Biotechnology Information b37.1 (NCBI). Reads that passed were then aligned to the human reference genome (UCSC hg19) using the Burrows-Wheeler Aligner (University of California, Santa Cruz, CA). Integrative Genomics Viewer software (Broad Institute, Cambridge, MA) was used for sequence data visualization. The base quality recalibration and local realignment were performed by the Genome Analysis Toolkit (Challis et al., 2012). Single nucleotide polymorphisms (SNPs) and small insertions/deletions were called with SAMtools (version 0.1.7). For copy number variations (CNVs), the CNsolidate, CoNVex, and CIFER were used, respectively, to detect them in the array CGH and exome data and to predict their inheritance (Wright et al., 2018). The variant calling (VCF file) containing these variants was annotated with Variant Effect Predictor v83 and the dbNSFP (Database for Nonsynonymous SNPs' Functional Predictions) v3.1.
The following bioinformatics pipeline was applied to narrow down the number of candidate diagnostic SNVs, indels, and CNVs (Wright et al., 2018): (1) variants with an allelic frequency >0.1% in the Exome Variant Server (EVS, http://evs.gs.washington.edu/EVS/), dSNP147 (http://ncbi.nih.gov/SNP), the 1000 Genomes Project (1000G, http://1000genomes.org/), and Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org) were disqualified and excluded; (2) variants predicted to be dysfunctional within a coding gene were eligible and retained, such as transcript ablation, transcript amplification, frameshift, splice donor and/or acceptor, stop gained and/or lost, start lost, in-frame deletion and/or insertion, and missense variants; (3) variants must overlap a confirmed or probable dRTA-related gene, and their corresponding genotypes need to meet the allelic requirement of the gene. For SNVs/indels, this includes single heterozygotes in dominant genes, homozygotes, and compound heterozygotes in recessive genes. For CNVs, this includes deletions and disruptive intragenic duplication, whole-gene/exon duplications, and any large (>1MB) genic deletions/duplications in dRTA-related genes. SNV/CNV compound heterozygotes were also evaluated in biallelic genes; (4) variants in the proband must be inherited in a manner that is both consistent with the family history of disease (assuming full penetrance) and the inheritance pattern of the gene (dominant/recessive), including inherited homozygous and compound heterozygous variants in recessive genes, inherited heterozygotes in dominant genes inherited from an affected parent, and de novo mutations in dominant; (5) exclusion of variants predicted to be benign by online tools (i.e., PolyPhen-2: http://genetics.bwh.harvard.edu/pph2/, SIFT: http://sift.jcvi.org/and MutationTaster: www.mutationtaster.org/); and (6) the novel variant was verified in the HGMD (http://hgmd.cf.ac.uk/ac/index.php) and ClinVar (http://ncbi.nlm.nih.gov/clinvar).
In silico analyses
Three online programs (i.e., PolyPhen-2, SIFT, and MutationTaster) were used to predict pathogenicity of the variants detected by the NGS analysis. On the basis of matrix algorithm described by Grantham et al., further score system evaluating the deleteriousness of missense mutation was used in this study. Moreover, for amino acid substitutions, multiple sequence alignments on 8 species for AE1, and subunit B1 of apical H+-ATPase through Vector NTI Advance 10-Align were used to evaluate evolutionary conservation. Those eight species for AE1 were as follows: mus musculus (NP_035533), Gallus gallus (NP_001277483), Xenopus tropicalis (XP_002935581), Callorhinchus milii (XP_007899713), Camelus ferus (XP_014423981), Pan troglodytes (XP_016787164), Lonchura striata domestica (XP_021394174), and human (XP_011523431); and those for submit B1 of apical H+-ATPase: Mus musculus (NP_598918), Rattus norvegicus (NP_001101337), Xenopus tropicalis (NP_001107734), Pan troglodytes (XP_001145161), Columba livia (XP_021136741), Lonchura striata domestica (XP_021382522), Delphinapterus leucas (XP_022416961), and human (NP_001683).
The Berkeley Drosophila Genome Project (BDGP, available at www.fruitfly.org) and the HSF 3.0 (Human Splicing Finder, available at www.umd.be/HSF3/) splice prediction programs were used to evaluate the effect of DNA variants on the exonic splicing process.
Sanger sequencing
Sanger sequencing was applied to the missed coverages by NGS in the probands, aiming to rule out the presence of other variants. Also, the potential candidate variants identified by NGS were validated by Sanger sequencing in five probands and their family members. The uncovered bases, suspected candidate mutations site, and its flanking regions were amplified by Prism 3700 DNA Analyzer (Applied Biosystems, CA). The PCR product was subcloned into the PGEM-T Easy vector (A1360; Promega) and sequenced using the T7/SP6 primers when heterozygous deletion or insertion was suspected.
Results
Clinical findings
We studied five Chinese patients with dRTA belonging to five unrelated kindreds. Four patients were first admitted in 0-3 months after birth due to failure to thrive (patient I and II), diarrhea, dehydration (patient III), and neonatal pneumonia (patient V), while patient IV was admitted due to growth delay and fatigue at the age of 4. As can be seen from Table 1, all patients had metabolic acidosis with normal serum anion gap (blood pH <7.35; average value of serum bicarbonate 9.9 ± 3.6 mmol/L), higher-than-normal serum chloride concentration (average value 117.8 ± 6.5 mmol/L), hypokalemia (average value 2.77 ± 0.37 mmol/L), and paradoxical alkali urine (UpH >6.0 while CO2CP <18 mmol/L). In addition, bilateral nephrocalcinosis and growth delay were shown in all patients. Among of them, two of five patients suffered rickets (patient I and IV). Regretfully, imaging examination of inner ear was not performed to determine the presence of enlarged vestibular aqueduct (EVA), although all patients passed the screening test of bilateral ears at birth, and none has an abnormal hearing currently. No significant metabolic acidosis, hypokalemia, or kidney stones were discovered in other family members of all the five patients (including those with heterozygous mutations).
Gene analysis
The NGS coverage parameters of five probands, including the genes of the ATP6V0A4, ATP6V1B1, SLC4A1, and FOXI1, were shown in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/gtmb). The coverage percentage of targeted exons of four genes at 20 × was 99.75-100%. Sanger sequencing showed negative results in the missed coverages using NGS in the FOXI1 of patient II and ATP6V1B1 of patient III.
As shown in the Table 2 and Figure 1, four different mutations of ATP6V0A4 gene were found in two patients (patient I and III) with dRTA by NGS: Patient I was a compound heterozygote with a novel intronic mutation of c.639 + 1G>A and a nonsense variant of c.580C>T (p.Arg194*); patient III carried two duplications (compound heterozygote) of ATP6V0A4. The duplication c.1504dupT led to a frame shift from the 502nd tyrosine that resulted in premature termination of the protein at codon 523 (p.Tyr502Leufs*22), leading to a truncated protein; the other duplication c.2351dupT led to a frame shift from the 785th phenylalanine that resulted in premature termination at codon 812 (p.Phe785Ilefs*28), leading to truncated 28 amino acids at the COOH-terminus. Above-mentioned 4 mutations were inherited from their mother and father, respectively, with cosegregation from phenotypes. According to HGMD and some relevant literatures, three mutations of ATP6V0A4 gene were identified as novel ones in this study, except for a previously reported mutation (c.580C>T) (Stover et al., 2002). Patient II was identified to carry two novel missense mutations (compound heterozygote) of ATP6V1B1 gene (c.409C>T, p.Pro137Ser; c.904C>T, p.Arg302Trp), which were inherited from his mother and father, respectively. Meanwhile, patient IV was revealed to harbor a previously reported heterozygous missense mutation of SLC4A1 gene (c.1765C>A, p.Arg589Ser) (Karet et al., 1998). However, no mutation was found in his parents, suggesting that c.1765C>A was de novo. No mutation of the dRTA-related genes was found in patient V. All the five novel mutations mentioned above were not found in the 100 healthy controls. Besides, none of the five patients was found with potential mutations of the FOXI1 gene.
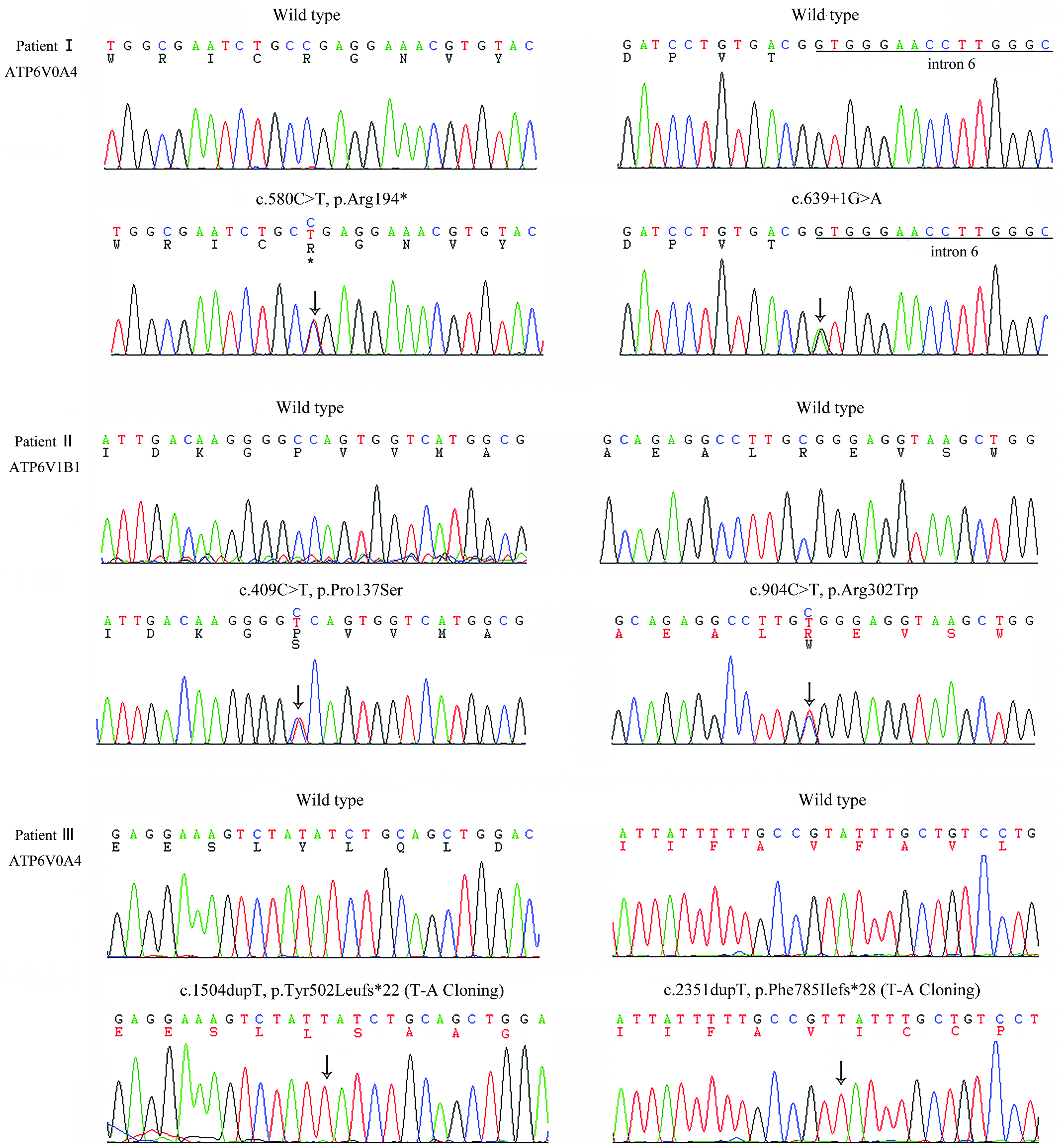
Novel mutations identified in the patients with dRTA. The black arrows indicate the location of the mutation site. dRTA, distal renal tubular acidosis. Color images available online at www.liebertpub.com/gtmb
Mutation naming and description rules refer to the latest guideline published by Human Genome Variation Society. a
Grantham Matrix scoring system and the three types of in silico software (SIFT, PolyPhen-2, and MutationTaster) were performed to predict the pathogenicity of the missense mutations, including two missense variants of ATP6V1B1 (c.409C>T, p.Pro137Ser; c.904C>T, p. Arg302Trp) and one of SLC4A1 (c.1765C>A, p.Arg589Ser) (Supplementary Table S2 and S3). It turned out that the Grantham Matrix scoring system and all the three online softwares predicted that both p. Arg302Trp and p.Arg589Ser were deleterious mutations with high probability. p.Pro137Ser was evaluated to be harmful with moderate probability by Grantham Matrix scoring system. However, p.Pro137Ser was also predicted as deleterious by two different softwares expect for the PolyPhen-2 with the prediction of possibly damaging (the score 0.544). Sequence alignment demonstrated that the sites of these three missense mutations (p.Pro137Ser and p. Arg302Trp of ATP6V1B1, and p.Arg589Ser of SLC4A1 gene) were highly conserved in all the eight species of their respective homologous proteins. In addition, none of the three missense mutations was reported in the databases of 1000G or ExAC.
It was of note that the c.639 + 1G>A was a nucleotide substitution, which was located at the first position of intron 6, suggesting that it was a variant of classical splicing donor site (DS). Meanwhile, the c.639 + 1G>A was also predicted as a splicing variant resulting in the disappearance of DS of intron 6 (the score decreases from 0.12 to 0, cutoff 0.1) by the program BDGP. Therefore, the c.639 + 1G>A probably produced varied productions (e.g., a truncated protein as a result of exon 6 skipping of ATP6V0A4 gene). However, we failed to further perform in vivo analyses because the patient I and her guardians refused to offer more blood samples. Besides, HSF 3.0 predicted that the rest of mutations had no significant effect on splicing progress.
Treatment and follow-up
All patients accepted the treatment with alkali therapy and potassium supplement (sodium citrate: potassium citrate: citric acid = 1:1:0.6) to improve the metabolic alkalosis and electrolyte disturbance. We adjusted therapeutic program according to the degree of recovery from growth retardation and electrolyte disturbance. Growth curves of these patients (including height and weight) and the starting time of medication intervention are shown in Figure 2. From the Figure 2, we know that the height and/or body weight of five patients were at least one standard deviation lower than the mean values. Fortunately, all patients achieved obvious recovery that increased their growth to almost normal range through regular therapy. No renal insufficiency or deafness was detected in any of the patients during a follow-up period up to 9-36 months.

The growth curves of five patients with dRTA. Arrowheads indicate the start of treatment. SD, standard deviation. Color images available online at www.liebertpub.com/gtmb
Discussion
With the rapid development of molecular genetics, localization of causative genes has offered reliable evidence for the accurate diagnosis of disease and improved the physicians' understanding of pathophysiology and pathogenesis of primary dRTA. Although physiological complications of distal tubular function have remained somewhat elusive until recently, it is now clear that acidification dysfunction of dRTA is associated with functional defects of at least three molecules expressed in the intercalated cells of the renal tubule, which are cytoplasmic carbonic anhydrase II, AE1, and H+-ATPase, respectively (Palazzo et al., 2017).
At present, a total of 10 novel mutations of dRTA were reported in the Chinese population. Meanwhile, we had analyzed mutations of the AD inherited and recessive inherited dRTA from China and found five novel variants consisting of one of SLC4A1, 2 of ATP6V0A4, and 2 of ATP6V1B1 gene mutations in previous studies (Shao et al., 2010; Gao et al., 2014). This study aimed to analyze the molecular defects of the five patients with dRTA, newly diagnosed in China, to enhance the cognition and to enrich the gene mutation database of the disease.
By gene analysis, we have identified four mutations of ATP6V0A4, including three novel variants in two patients (patient I and III) with dRTA. All the above-mentioned mutations were pathogenic, which may result in exon skipping because of the weakness of splicing sites (c.639 + 1G>A), or lead to an unable mRNA due to the nonsense-mediated mRNA decay (p.Arg194*), or cause translational frame shifting and truncated proteins (c.1504dupT and c.2351dupT). Noteworthily, p.Arg194* in the state of homozygote had been described in a dRTA patient with early SNHL by our previous research (Gao et al., 2014), but patient I carrying the p.Arg194* in this study had normal hearing at present. The discrepancy with respect to the absence or late onset of SNHL in patients with mutations of A4 subunit is likely associated with polymorphisms and regulatory differences of modifying genes. In addition, the mutation c.639 + 1G>A in another allele of the patient might have a different influence on the disruption of 5′ splice site recognition in ATP6V0A4 gene in different organs or cells, and ultimately resulted in different phenotypes.
We had also identified two novel compound heterozygous mutations of ATP6V1B1 gene in the patient II: c.409C>T (p.Pro137Ser) and c.904C>T (p.Arg302Trp). The mutation p.Arg302Trp was predicted to be highly pathogenic by the Grantham Matrix scoring system and three online softwares. However, p.Pro137Ser was predicted to be moderate pathogenic by Grantham Matrix scoring system and PolyPhen-2, only with high pathogenicity in MutationTaster and SIFT. These two missense mutations could cause substitutions of highly conserved amino acids of B1 subunit in eight species, possibly leading to reduced or disabled B1 subunit. The relatively weak pathogenicity of the p.Pro137Ser might explain why the inner ear phenotype in patient II was relatively mild (without early-onset SNHL). Of course, further research is strongly needed to confirm its pathogenesis. Patient IV was identified to harbor a previously described heterozygous missense mutation (c.1765C>A, p.Arg589Ser) of SLC4A1 gene. Besides, the Arg509 is a hotspot mutation site, which is located in the intracellular domain between the sixth and the seventh transmembrane regions of the AE1 protein, and two other forms of mutations in the same site (p.Arg589His, p.Arg589Cys) have been confirmed to be pathogenic (Bruce et al., 1997; Jarolim et al., 1998; Karet et al., 1998).
In this study, no dRTA-related mutation was found in the patient V, which may be due to the fact that it was difficult to find the large rearrangement with multiple exons involved in, or mutations undetected by this method were located in deep intronic segments, or there was the existence of novel dRTA loci. About one-third of patients with a clinical diagnosis of primary dRTA have no identified related mutations in currently known disease causative genes, suggesting that there are still unknown pathogenic genes to be found and thus whole-exome sequencing is more economic and feasible than Sanger sequencing (Enerbäck et al., 2017; Palazzo et al., 2017). Recently, the finding that the mutations in the FOXI1 gene caused dRTA confirmed this speculation of genetic heterogeneity (Enerbäck et al., 2017). Further experiment with larger panel for detection of potential novel dRTA loci was strongly needed, however, we are unable to get enough economic support currently now.
As shown in the Table 1 and 2, all 5 dRTA patients involved in our research carried different genetic spectrums with each other, although they manifested with similar clinical features, making it hard to figure out their association of genotype-phenotype. Some published data have found that the dominant form of dRTA caused by the mutation of SLC4A1 typically presents in adolescence or adulthood, whereas recessive variants caused by the mutation of ATP6V0A1/ATP6V1B1 occur in infancy or early children (Bruce et al., 1997; Khositseth et al., 2012; Palazzo et al., 2017). Furthermore, the age at clinical diagnosis in the dRTA patient with mutation of the SLC4A1 gene (patient IV) was significantly older than that of three patients (patient I, II, and III) carrying mutations of the ATP6V0A4/ATP6V1B1 gene in this study, which was consistent with previous studies.
SNHL is an important clinical feature in the dRTA patients carrying the mutations in the ATP6V0A4/ATP6V1B1 gene. It has been typically believed that loss-of-function mutations in the ATP6V1B1 gene cause dRTA with early SNHL, whereas ATP6V0A4 mutations are classically associated with either late onset of SNHL or normal hearing (Vargas-Poussou et al., 2006). Recently, V Palazzo et al. (2017) reported that 92% of patients were found to have SNHL in the group of patients harboring causative mutations in the ATP6V1B1 gene, and 56.7% with pathogenic variants in the ATP6V0A4 gene had SNHL, which was slightly higher than that reported previously. Interestingly, patients with ATP6V0A4 mutations presented a wide range of clinical onset time of SNHL, encompassing infancy and early childhood. Surprisingly, V Palazzo et al. (2017) first reported a dRTA case presented SNHL with mutations of SLC4A1 gene, which may be associated with the presence of biallelic mutations-induced deafness or the pervasiveness of hearing loss in the population.
Moreover, studies have confirmed that the EVA syndrome is relevant with mutations in the ATP6V0A4 and ATP6V1B1 gene (Joshua et al., 2008; Lorente-Cánovas et al., 2013; Gao et al., 2014). The EVA, as an anomaly of the inner ear structure, might lead to SNHL or mixed hearing loss (HL) and manifest with normal hearing, congenital total deafness, progressive HL, or fluctuating HL (Gao et al., 2014). Unlike the general pattern, none of the patients in this study with ATP6V1B1/ATP6V0A4 gene had an abnormal hearing, especially in patient II who carried two heterozygous missense mutations of ATP6V1B1, which may be related to the slight impact of variant B1 subunit on the inner ear or modification of relevant genes in their younger age. A drawback of this study was lacking of imaging studies to confirm the existence of the EVA and careful audiological testing to assess hearing, which hindered the in-depth analysis of the association between audiologic phenotype and genotype. Therefore, detection of hearing changes and imaging studies are necessary for the dRTA patients during follow-up.
Of note, five patients in this study suffered from obvious growth retardation. Fortunately, all patients obtained a significant improvement in delayed growth, and their serum potassium returned to normal levels after timely and accurate diagnosis and treatment. In some patients with a long history of dRTA, chronic renal insufficiency may occur because of progressive tubulointerstitial damage resulted from nephrocalcinosis and persistent hypokalemia (Palazzo et al., 2017). This suggests that patients with the typical/atypical dRTA should be diagnosed and treated early to prevent irreversible complications, especially in patients with nephrocalcinosis, whose renal function should be closely followed up.
In conclusion, we have found seven mutations in three different genes associated with dRTA, including one mutation in SLC4A1 gene, two in ATP6V1B1 gene, and four in ATP6V0A4 gene. Among them, five ones were novel, which further expanded the mutation database of dRTA. This investigation not only deepened the understanding of the molecular pathogenesis of the dRTA but also provided reliable reference data for genetic counseling and treatment.
Footnotes
Acknowledgments
We are grateful to all subjects for their participation. This study was supported by grants from the Natural Science Foundation of China (81170653) and the Shandong Provincial Natural Science Foundation (ZR2014JL054).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.