
Abstract
Background:
Mucolipidosis III gamma (MLIIIγ) is a rare autosomal recessive disorder characterized by radiographic evidence of mild-to-moderate dysostosis multiplex, progressive joint stiffness and pain, scoliosis, and normal to mildly impaired cognitive development. Cardiac valve involvement and respiratory complications can be significant. MLIIIγ is caused by mutations in the GNPTG, which encodes the γ subunit of the enzyme N-acetylglucosamine-1-phosphotransferase.
Objective:
Clinical and genetic study of seven individuals of a consanguineous Pakistani family affected with mucolipidosis phenotype who never pursued medical care.
Methods:
Genome-wide homozygosity mapping was performed using Affymetrix Human SNP Array 6.0 followed by whole exome and Sanger sequencing.
Results:
The affected individuals showed characteristic clinical features of MLIIIγ. Whole-genome single nucleotide polymorphism genotyping identified a region of homozygosity shared by affected individuals of the family on chromosome 16p13.3. Whole exome sequencing identified a novel 4-bp deletion in the GNPTG segregating in the family in agreement with autosomal recessive pattern.
Conclusions:
We identified a novel mutation in the GNPTG gene as the underlying cause of MLIIIγ in a Pakistani family. This study supports the role of next-generation sequencing technologies for the molecular diagnosis of rare inherited disorders.
Introduction
M
GlcNAc-1-phosphotransferase is a heterohexameric complex made up of three subunits: 2α, 2β, and 2γ (Bao et al., 1996). The α and β subunits of GlcNAc-1-phosphotransferase (encoded by GNPTAB on chromosome 12q23.3) harbor the catalytic site and a domain that mediates the specific recognition of the acid hydrolase (van Meel et al., 2016). The γ subunit of GlcNAc-1-phosphotransferase is a glycoprotein (encoded by GNPTG on chromosome 16p13.3) and enhances the phosphorylation of a subset of the lysosomal acid hydrolase substrates (Qian et al., 2010).
Mutations in GNPTAB may either cause a severe lysosomal storage disorder MLII (I-cell disease) or an attenuated phenotype MLIIIα/β (pseudo-Hurler polydystrophy) depending on the GLcNAc-1-phosphotransferase activity. Mutations in GNPTG result in the least severe phenotype MLIIIγ. However, MLIIIα/β and MLIIIγ are clinically indistinguishable. The patients typically have short stature, claw-hand deformity, stiffness of the finger and shoulders, scoliosis, valvular heart disease, and sometimes mild mental retardation. The symptoms appear in the early childhood and continue in a slowly progressive pattern throughout life (Braulke et al., 2013).
In this study we report clinical and genetic study of a large consanguineous Pakistani family affected with mucolipidosis phenotype and identified a novel frameshift mutation in GNPTG through next-generation sequencing.
Methods
Human subjects
We ascertained a large consanguineous Pakistani family affected with MLIIIγ living in a remote village of Charsadda District in Khyber Pakhtunkhwa Province (Fig. 1). They were clinically investigated at the Leady Reading Hospital, Peshawar. Before the start of the study, approval was obtained from the Quaid-i-Azam University Institutional Review Board. Informed consent was obtained from all the family members who participated in the study. Venous blood samples were collected from six affected (III-15, III-18, IV-1, IV-2, IV-7, and IV-8) and five normal individuals (III-5, III-6, III-8, III-14, and IV-12) in EDTA tubes and genomic DNA was extracted from whole blood following a standard protocol.
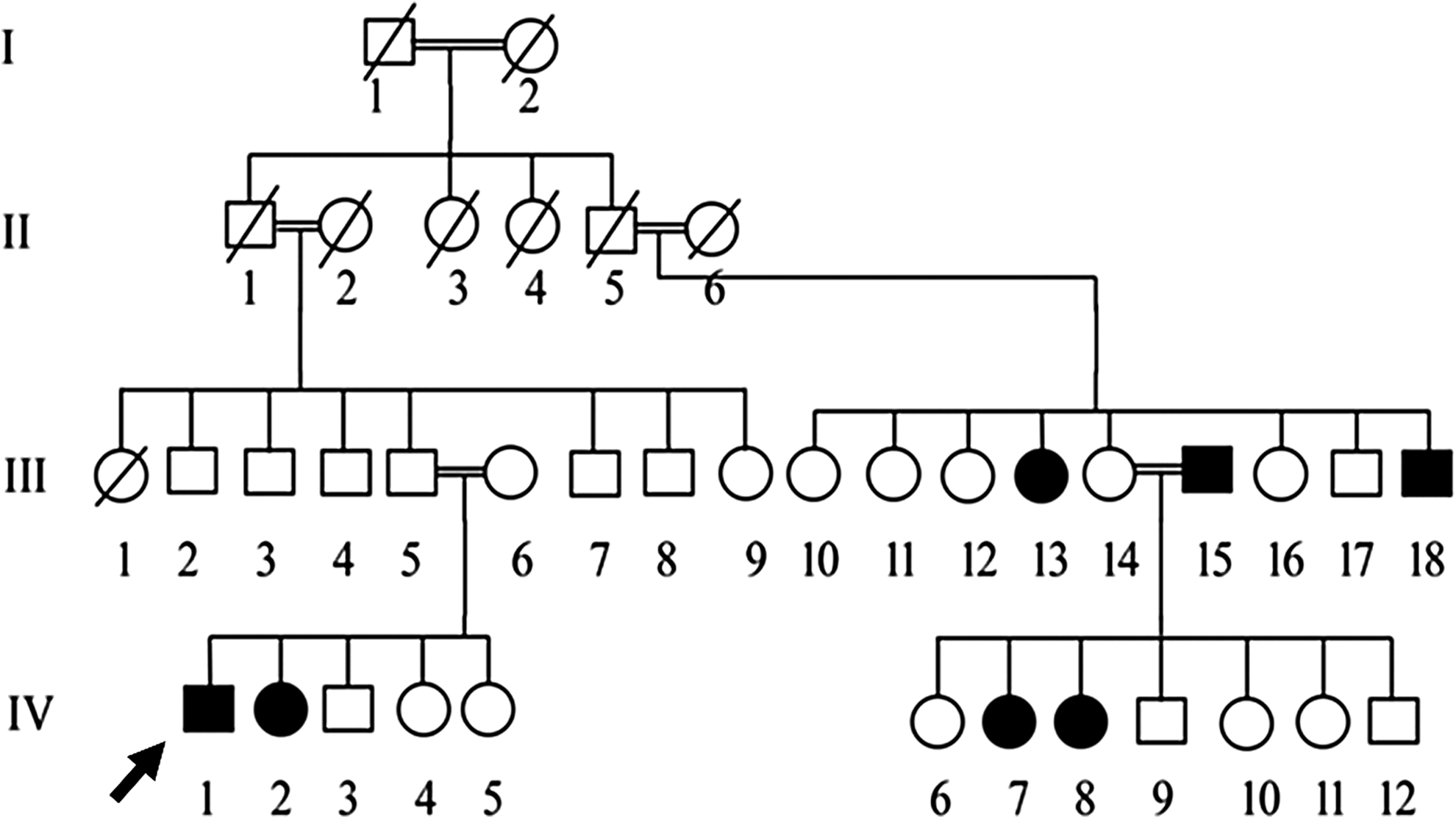
A four-generation pedigree of the family affected with MLIIIγ enrolled in this study. The proband is indicated with an arrow. MLIIIγ, mucolipidosis III gamma.
Genome-wide SNP genotyping
gDNA samples of the affected and normal individuals of the family were subjected to SNP microarray (Genome-Wide Human SNP Array 6.0; Affymetrix, Santa Clara, CA). The array data were genotyped with a genotyping data analysis program (Genotype Console, ver. 2.1, Affymetrix), and regions of homozygosity were identified using homozygosity mapper (Seelow et al., 2009).
Whole exome sequencing
Whole exome sequencing of patient IV-2 was performed using SureSelect V4 kit for target enrichment followed by sequencing of the libraries through HiSeq2000 that gave 57,705,494 reads with an average size of 101 bp. BWA (bwa-0.7.10), Picard (picard-tools-1.1180), GATK (GATK3.v4), and SnpEff (SnpEff_v4.1) were used to map the reads to reference sequence, mark duplicates, variant calling, and variant annotation, respectively, reducing number of variants to 63,036.
Bioinformatic analysis
Based on autosomal recessive mode of inheritance of the disease phenotype in the family, we filtered candidate variants using the following criteria: (1) homozygous or compound heterozygous in the affected individuals, and heterozygous in obligate carriers for the mutant allele; (2) present in coding exons or splice junctions; (3) nonsynonymous, frameshift, gain/loss of stop codons; (4) having a functional effect, for example, predicted to be pathogenic or deleterious by in silico prediction software including Polyphen-2 and SIFT; (5) minor allele frequency (MAF) <0.01 in the 1000 Genomes Project database (www.1000genomes.org/); and (6) not present in in-house exome sequence data obtained from 30 unrelated individuals.
Sanger sequencing
Segregation of the shortlisted variants with the disease phenotype in the family was tested through Sanger sequencing. Polymerase chain reaction (PCR) amplification of the genomic region flanking the potential candidate GNPTG variant was performed with the primers 5′-GGAGATCGCCAACAACACCTTC-3′ (forward primer) and 5′-ACCCCAAGCTGTCAGGACCTC-3′ (reverse primer). The PCR products were purified using Thermo Scientific GeneJET PCR Purification Kit (#K0702) and subsequently subjected to sequencing by Applied Biosystems automatic sequencer 3730xl using Big Dye terminator cycle sequencing kit V3.1 (PE Applied Biosystems, Foster City, CA). The sequences generated were analyzed in comparison with the reference sequence using BioEdit Sequence Alignment Editor (Version 7.0.5.3).
Results
Clinical characteristics of the patients
We recruited a large consanguineous family with seven affected individuals in two generations (Fig. 1). The family never pursued medical care before this study. The affected individuals had onset of skeletal symptoms at the age of 3 years. They began complaining of joint stiffness and loss of flexibility, which presented contractures of the hand and progressive joint stiffness, especially in the fingers, wrist, hip, and knees, and typical claw hands (Fig. 2). All affected individuals of the family complained of difficulty with walking after 9 years. Patients III-13 and IV-8 had serious hip pain. Radiographic evaluation of all patients showed signs of the spondyloepiphyseal dysplasia (Fig. 3). Echocardiography showed thickening of aortic valve in one affected individual (III-13) only. Pulmonary function tests and abdominal ultrasonography did not show any abnormality. The intelligence in all patients was normal. Four affected individuals (III-13, IV-1, IV-2, and IV-7) showed coarse facial expression (Fig. 2) and corneal clouding as well.
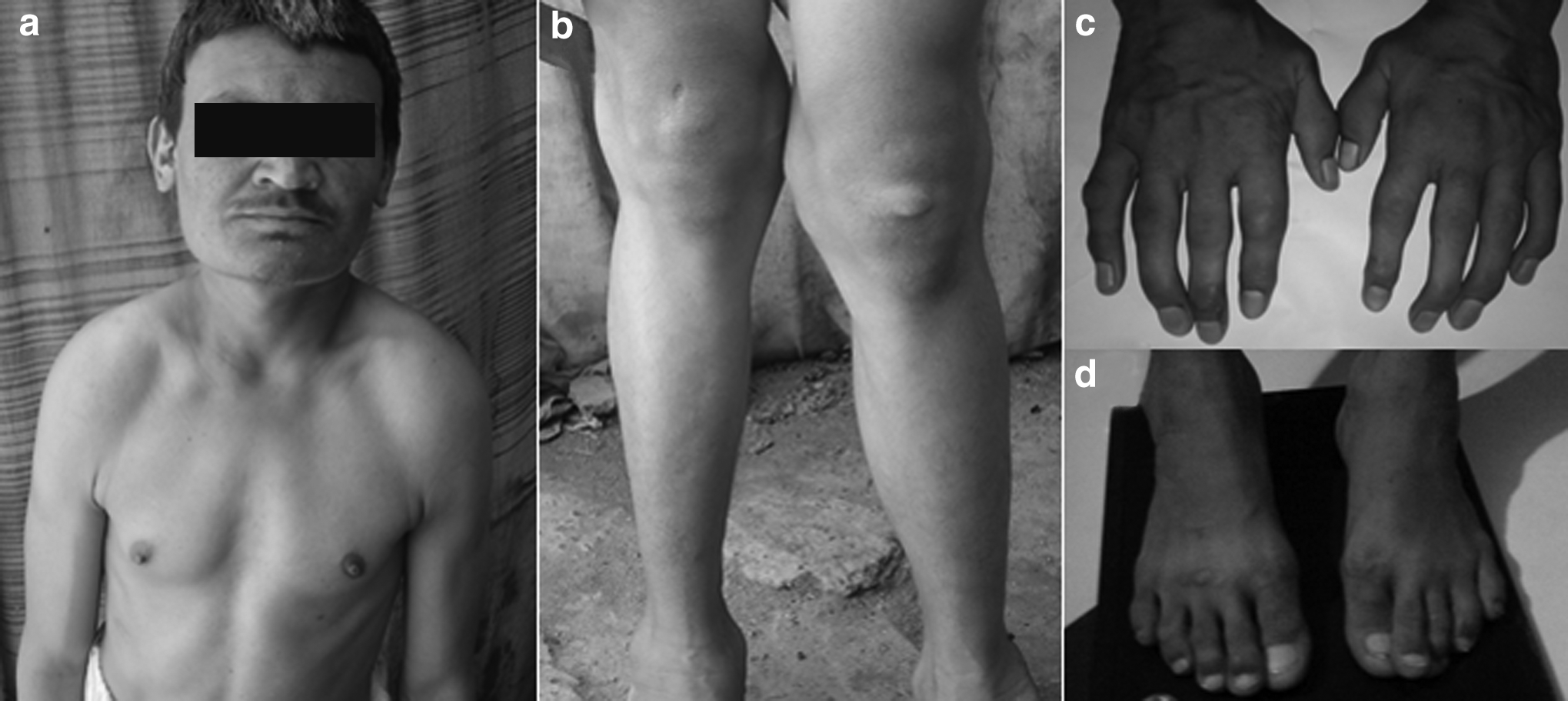
Clinical presentation of the affected individual IV-1: coarse facial features

Radiographs of the affected individuals showing signs of spondyloepiphyseal dysplasia.
Genome-wide SNP genotyping
We detected two long runs of homozygosity shared among affected individuals on chromosomes 10 and 16 (Fig. 4). However, the homozygosity on chromosome 10 was also shared by the normal individuals of the family, suggesting that homozygosity on chromosome 16 was identity by descent in the family. The mapped region harbored 240 protein coding genes. The prime candidate gene could be the GNPTG, but we decided to embark on whole exome sequencing of one affected individual IV-2 to rule out the possibility of any other comorbid mutations.

Homozygosity mapping from chromosomes 1 to 22 (left to right). The bars pointed by arrows represent the homozygous regions on chromosomes 10 and 16, respectively. The homozygosity scores are given along the Y-axis, relative to genomic position on the X-axis.
Whole exome sequencing
On average, 97% of read bases showed a Phred score >20 and the total number of bases in the reads was 5.7 Gb. The coverage of the targeted regions (more than 1 × ) was 98.8%, whereas the mean depth of targeted regions was 58.1 × . A total of 63,036 variants were found from whole exome and met quality control criteria (minimum depth >10 and minimum Q call >20). These were missense, nonsense, silent, insertion, and deletion variants.
The list of identified variants was narrowed down after excluding those that did not show a recessive mode of inheritance, those with an MAF >0.01 based on the 1000 Human Genome Database, those causing neutral or no functional effects based on in silico analyses to predict pathogenic effect, and those that were not present in our in-house exome sequence data of an unrelated healthy Pakistani population (n = 30). As a result, three candidate variants were shortlisted: GNPTG p.A160fs (NM_032520, c.478_479insTAGG), MRPS34 p.Q107fs (NM_023936, c.320_321), and PKD1L2 p.Q658X (NM_001076780, c.C658T). Sanger sequencing of these three variants in all family members verified that c.478_479insTAGG is the likely cause of mucolipidosis phenotype in this family: (1) this variant lies within the region of homozygosity (identity by descent) on chromosome 16 identified through whole-genome SNP genotyping and (2) only this variant cosegregated with the phenotype in recessive mode of inheritance in the family. All affected individuals (III-15, III-18, IV-1, IV-2, IV-7, and IV-8) were homozygous, whereas normal individuals (III-5, III-6, III-8, III-14, and IV-12) were heterozygous for the mutant allele (Fig. 5).
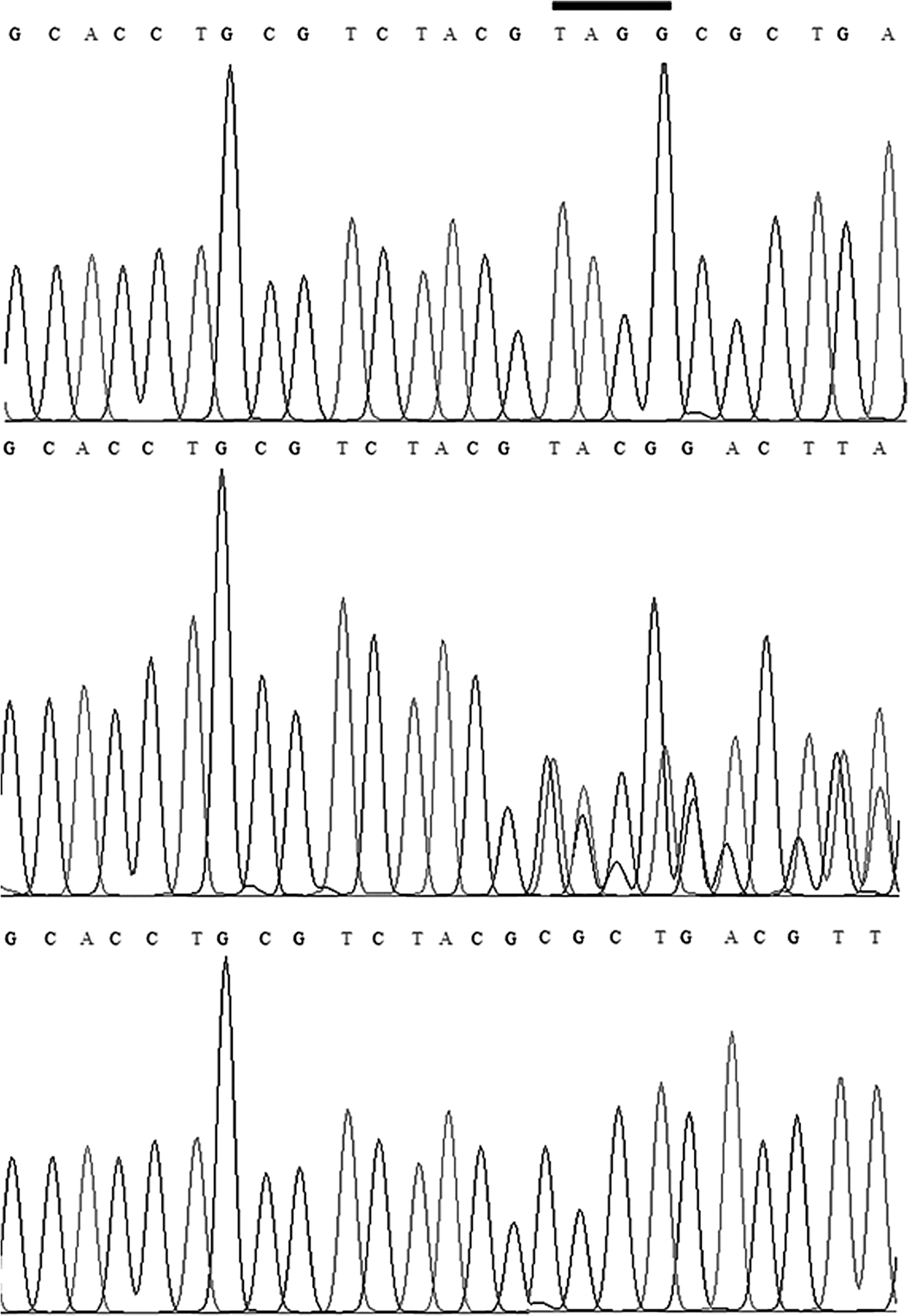
Sequence chromatograms showing 4-bp insertion mutation in the gDNA sequence of GNPTG amplified from the affected individuals of the family (top panel), heterozygous carriers (middle panel), and control individuals without mutant allele (bottom panel). The 4-bp insertion sequence is indicated with a bar in the top panel.
Discussion
We ascertained a large consanguineous Pakistani family segregating MLIIIγ in autosomal recessive mode of inheritance. Genome-wide SNP genotyping provided evidence of linkage to chromosomes 10 bearing 240 protein coding genes. Whole exome sequencing found an insertion mutation c.478_479insTAGG in GNTPG, which cosegregated with the disease phenotype in the family. Forty-two GNPTG mutations have been reported so far, including missense (18), nonsense (3), deletions (9), insertions (6), and splice site (6) mutations, randomly distributed in all exons of the gene (www.hgmd.cf.ac.uk).
The insertion c.478_479insTAGG predicts a frameshift and incorporation of a premature stop codon at position 199 so that the mutation fits in group of “pathogenic very strong 1” (PVS1) according to ACMG Standards and Guidelines (Richards et al., 2015). In a previous study, fibroblasts were obtained from two affected individuals identified with a frameshift GNPTG mutation (p.V176GfsX18), leading to a predicted truncated protein of 194 amino acids, and cell extracts were analyzed through Western blot and real-time PCR (Pohl et al., 2010). Both the mutant GNPTG mRNA and the possibly synthesized truncated protein were not detected, supporting activation of nonsense-mediated mRNA decay (Maquat, 2004). We correlate that frameshift mutation (c.478_479insTAGG) identified in our patients might lead to reduced levels of the γ subunit of GlcNAc-1-phosphotransferase complex due to nonsense-mediated mRNA decay, preventing the synthesis of the truncated protein p.(Ala160ValfsTer40). However, expression study of this novel mutation is indicated to explore the underlying mechanism of pathogenicity.
The studied family was highly inbred and all normal individuals who participated in the study were identified as heterozygous carriers for the mutant allele. Genetic counseling was provided to the family indicating risk of segregation of homozygous mutant allele through consanguineous marriages.
Conclusion
We reported a novel mutation in the γ subunit of N-acetylglucosamine-1-phosphotransferase enzyme. The study led to carrier identification and genetic counseling of the affected family and should be helpful in prenatal diagnosis.
Footnotes
Acknowledgments
We are grateful to the family members for participating in the study. M.A.K. is supported by Higher Education Commission of Pakistan through Indigenous PhD Fellowship (PIN: 117-1632-BM7-266).
Author Disclosure Statement
No competing financial interests exist.