
Abstract
Aims:
To identify potential causative gene mutations in a large Han Chinese pedigree with diffuse nonepidermolytic palmoplantar keratoderma (NEPPK).
Methods:
We enrolled 11 patients and 8 healthy individuals from a pedigree with NEPPK and 100 randomly selected healthy controls. Biopsy samples were obtained from the proband. Genomic DNA was extracted from a peripheral blood sample from each participant. Mutation detection via polymerase chain reaction and Sanger sequencing of relevant potential causative genes, including KRT1, KRT6C, KRT10, KRT16, AQP5, and SERPINB7, was performed. Comparisons were made between sequencing outcomes and currently available reference genome databases, including HGMD Pro, Pubmed, 1000 Genomics, and dbSNP.
Results:
Histological findings, clinical features, and medical history were in accordance with the diagnosis of diffuse NEPPK. We identified a novel splice-site mutation c.1255-1G > C in intron 6 of KRT1 in all individuals with NEPPK in the pedigree.
Conclusions:
Diffuse NEPPK is a relatively rare subtype of palmoplantar keratoderma. The results of this study expand the spectrum of KRT1 mutations in diffuse NEPPK and provide insights into the understanding of its underlying pathological mechanisms and phenotype-genotype correlations.
Introduction
Palmoplantar keratoderma (PPK) is a type of highly heterogeneous genodermatoses, with primary clinical features of hyperkeratosis of thick and yellowish skin with a sharp demarcated border at palms and/or soles. PPKs comprise hereditary and acquired conditions, and hereditary PPKs can be classified following different approaches. Based on different clinical features, hereditary PPKs can be divided into isolated PPKs, nonsyndromic PPKs with additional cutaneous manifestations, and syndromic PPKs with extra cutaneous features (Guerra et al., 2018). Based on variant palmoplantar phenotypic patterns, PPKs can be further classified into diffuse PPKs, focal PPKs, striate PPKs, punctate PPKs, etc. In addition, histological findings may further distinguish epidermolytic PPK from nonepidermolytic PPK.
Hereditary diffuse PPK is also known as keratoderma palmoplantar hereditaria or Thost-Unna syndrome, which can be inherited from parents with a causative gene mutation, and is the most common pattern of hereditary isolated PPKs. This condition has a low prevalence worldwide, although the precise figures on the incidence and prevalence are not available, as many patients with mild or atypical PPKs do not seek medical care (Schiller et al., 2014; Has and Technau-Hafsi, 2016).
Based on histological patterns, hereditary diffuse PPKs can be classified into diffuse nonepidermolytic palmoplantar keratoderma (NEPPK, OMIM #600962) and diffuse epidermolytic palmoplantar keratoderma (EPPK, OMIM #144200). KRT9 is the predominant causative gene for EPPK, whereas genetic defects in KRT1 may mainly attribute to NEPPK (Kimonis et al., 1994). Human NEPPKs exist in special forms, which include focal forms, diffuse Bothnian types, and diffuse Nagashima types, and are caused by variant genes, such as KRT16, AQP5, SERPINB7, DSG1, and KRT6C (Smith et al., 2000; Keren et al., 2005; Blaydon et al., 2013; Kubo et al., 2013; Wang et al., 2018). Rogaev et al. (1993) detected a mutated KRT10 in a patient with NEPPK cosegregated with phenotypes, indicating heterogeneity of diffuse NEPPK.
In this study, we evaluated a large four-generation Han Chinese pedigree affected by diffuse NEPPK and performed sequencing on possibly causative genes, including KRT1, KRT6C, KRT10, KRT16, AQP5, DSG1, and SERPINB7. We aimed to further expand the spectrum of causative gene mutations in diffuse NEPPK and provide additional information to better understand the role of KRT1 in the pathogenesis of diffuse NEPPK.
Materials and Methods
Ethical approval
The Ethics Committee of the Second Affiliated Hospital of Xi'an Jiaotong University approved this study protocol (No. 2AH_XJTU_2018207), and all clinical procedures were conducted strictly according to the principles of the Helsinki Declaration.
Participants
We enrolled a large four-generation Chinese pedigree that consists of 28 individuals from the Shaanxi Province of the People's Republic of China (Fig. 1). After obtaining informed consent, detailed clinical information and images of cutaneous lesions were collected from 11 individuals with NEPPK from the pedigree (II:2, II:5, II:7, III:2, III:3, III:6, III:7, III:10, IV:1, IV:3, and IV:4). In addition, 100 unrelated ethnically matched healthy controls were randomly enrolled (male/female: 50/50; mean age: 25.4 ± 3.8 years).

Pedigree of family with 28 members, among which 13 individuals were diagnosed with diffuse nonepidermolytic palmoplantar keratoderma; arrow indicates the index patient.
Histological studies
Biopsy samples from cutaneous lesions on the left palm of the proband (III:2) were obtained. Hematoxylin-eosin staining, slice making, and pathological examination were then performed.
Genetic studies
Peripheral blood samples were obtained from the 11 individuals with NEPPK, 8 healthy pedigree members, and 100 controls. Genomic DNA was extracted from leukocytes according to the instructions of the TIANamp Genomic DNA Kit (Tiangen Biotech, China). Primers flanking all the exons and adjacent introns of NEPPK-relevant genes, including KRT1, KRT6C, KRT10, KRT16, AQP5, DSG1, and SERPINB7, were designed using the Primer 5.0 program. Polymerase chain reaction (PCR) assays of all targeted genes were conducted as previously described (Tang et al., 2018). Sanger sequencing of the purified PCR products was performed using an ABI3730XL DNA analyzer (Applied Biosystems, CA). All sequencing outcomes were compared with currently available databases, including HGMD Pro, Pubmed, and dbSNP databases. Mutation Taster, a bioinformatical software program, was used to analyze the potential alteration of splice sites and the probable impact of the genetic variance of the translated protein.
Family members of the pedigree did not provide additional blood samples for subsequent testing, including real-time PCR aimed at elucidating the expression levels of targeted genes with mutations.
Results
Clinical data and histological findings
The proband (III:2) of the pedigree was a 37-year-old woman who presented with diffuse hyperkeratosis on both her palms and soles shortly after birth; however, there was no history of skin fragility or blistering. The yellowish cutaneous lesions were finely demarcated and skin creases displayed several deep fissures; the wrists and the dorsal aspects of the extremities were not affected (Fig. 2a, b). All the individuals with NEPPK in the pedigree displayed mild-to-severe hyperkeratosis confined to the palms and soles, which was similar to the index patient. Several patients in the pedigree experienced mild limitation of extension of digits, hyperhidrosis, and hangnails. Histological examination of multiple sections on the index patient revealed orthokeratotic hyperkeratosis, acanthosis, hypergranulosis, and mild lymphocyte infiltrations in the upper dermis; there was no evidence of epidermolysis (Fig. 2c). Detailed clinical features of the patients are shown in Table 1.
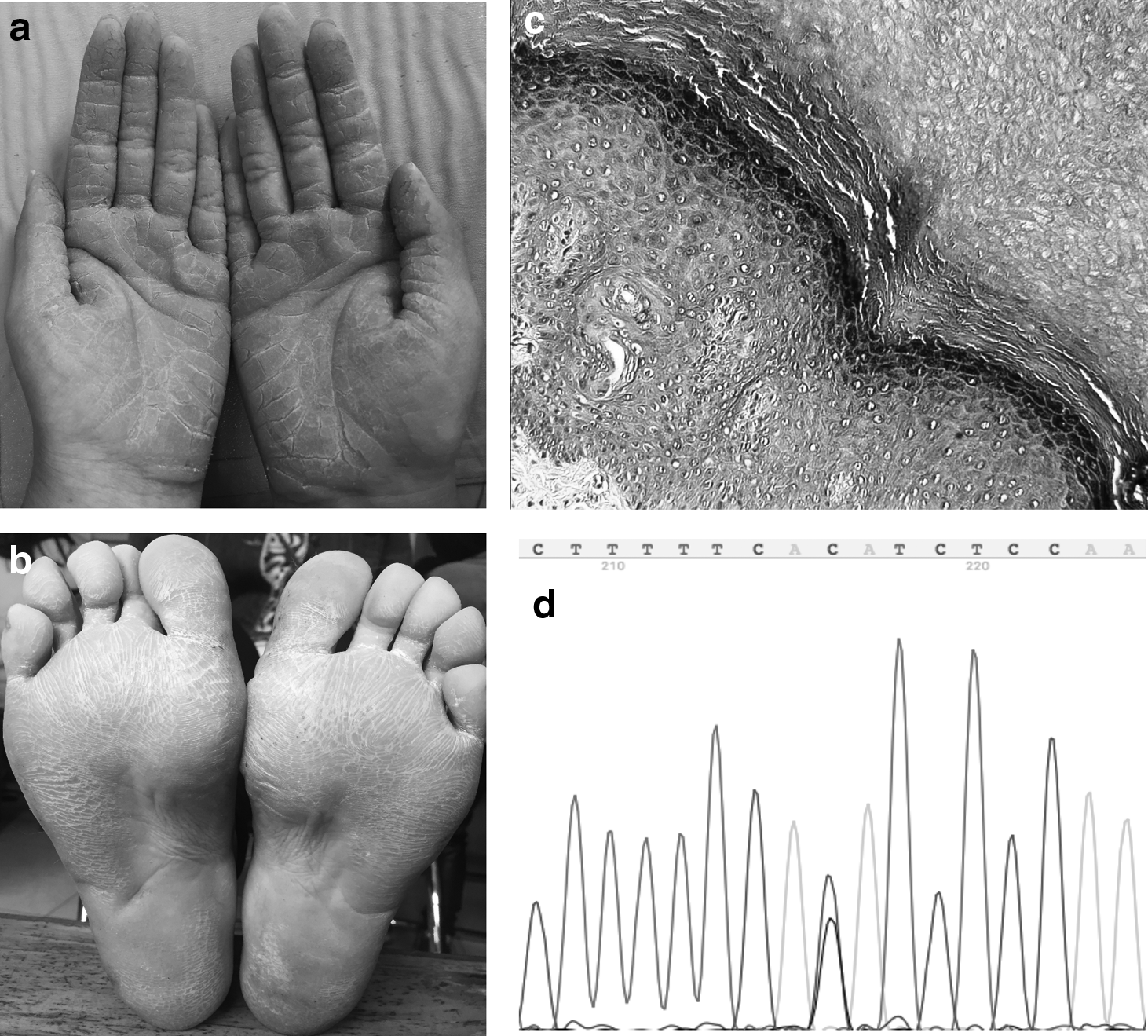
Well-demarcated skin lesions with an erythematous border on the palms and soles of the index patient
Clinical Features of Family Members with Nonepidermolytic Palmoplantar Keratoderma
Genetic study outcomes
Sanger sequencing of KRT1 revealed a novel splice-site mutation, c.1255-1G > C in intron6, in all individuals with NEPPK (Fig. 2d). This variant was not identified in the controls in the pedigree or in the 100 healthy controls. This gene mutation has not been previously reported and is not a known single nucleotide polymorphism based on public databases, including HGMD Pro, Pubmed, 1000 Genomics, and dbSNP. No mutations were identified in KRT6C, KRT10, KRT16, AQP5, DSG1, or SERPINB7 in any participants in this study.
The prediction outcomes of the novel splice-site mutation indicated a probable alteration in the splice-site position and the genetic defect is likely damaging (Fig. 3).

Predicted alteration within the mutated splice site. The detected mutation is likely to disturb normal splicing through direct loss, increase, marginal increase, or new gain of splicing acceptors.
Discussion
PPK is an umbrella term for any form of persistent thickening of the epidermis, mainly restricted to palms and soles, that includes hereditary and acquired patterns (Guerra et al., 2018). Diffuse PPKs consist of a multitude of disease entities accompanied by several cutaneous and extracutaneous clinical features. Diagnoses of different entities of diffuse PPKs are based mainly on age of onset, inheritance pattern, morphology, distribution of the skin lesions, and histological findings from biopsy samples.
NEPPK consists of numerous heterogenetic conditions that display various cutaneous lesions or distributions. Kimonis et al. (1994) first mapped the causative gene of diffuse NEPPK to 12q11-12q13, and identified a missense mutation in KRT1, which segregated completely with the disease. Located on chromosome 12q13.13 including nine exons in total, KRT1 encodes KRT1, which is putatively coexpressed with KRT9 and KRT10 in the epidermis of palms and soles and is the expression partner of KRT10 in nonglabrous skin (NCBI GenBank Database; Terron-Kwiatkowski et al., 2002).
KRT1 consists of 644 amino acids and is a member of the type II cytokeratins family, which includes basic or neutral proteins that are arranged in pairs of heterotypic keratin chains coexpressed during differentiation of simple and stratified epithelial tissues. KRT1 also belongs to the intermediate filament family and is specifically distributed in spinous and granular layers of epidermis with the same family member KRT10. Mutations in genes encoding these proteins may also attribute to epidermolytic hyperkeratosis (OMIM #113800) and ichthyosis with confetti (OMIM #609165) (Guerra et al., 2015).
In this studied pedigree, a splice-site mutation c.1255-1G > C in intron6 of KRT1 was detected in all individuals displaying phenotypic features of NEPPK. This mutation has not yet been listed in Human Intermediate Filament databases or other currently available databases and is likely the first splice-site mutation detected in NEPPK. Mutations in the adjacent regions of exons and introns are prone to elimination or alter the splicing acceptor site, thus changing the sequence or reducing the stability of mature mRNA (Piasecka et al., 2015). In this case, the splice-site mutation may lead to functional defect of KRT1 owing to aberrant expression and translation through haploinsufficiency pattern. The bioinformatic analysis indicated that the detected novel mutation is likely to disturb the normal procedures of genetic splicing. The transition of the nucleotide may directly lead to the loss of the splicing acceptor, hence altering the sequence and stability of mature mRNA.
The detected splice-site mutation lies in the 2B helix domain of KRT1, which is thought to be critical for keratin intermediate filament nucleation (Wu et al., 2000). This domain of the translated protein interacts with the distal helix of KRT10 and forms a parallel coiled-coil heterodimer with a predominantly hydrophobic intermolecular interface. Genetic mutations, which are detected in epidermis keratins mainly resulting in cutaneous diseases, lie predominantly in the helix 1A and 2B domains of this gene (Bunick and Milstone, 2017). Cases reporting gene mutations in patients with diffuse NEPPK are limited, and the underlying correlations between phenotype and genotype are not yet clear, as the severity of the disease may vary significantly according to different locations where gene mutations lie.
Conclusion
This study identified a novel heterozygous splice-site mutation, c.1255-1G > C, located in intron6 of KRT1 in a large Han Chinese family affected by diffuse NEPPK. These findings further expand the spectrum of gene mutations in diffuse NEPPK and elucidate the underlying role that different domains of KRT1 play in diffuse NEPPK.
Footnotes
Acknowledgments
We sincerely thank our colleagues and friends who helped us with the study. In addition, we would like to specifically thank all participants for their participation.
Author Disclosure Statement
No competing financial interests exist.