
Abstract
Aims:
Single nucleotide polymorphisms (SNP) can be used as genetic markers and for risk assessment of allele-linked diseases, which can provide information for clinical diagnosis. Large-scale microarray and next-generation sequencing methods have made genome-wide SNP genotyping possible. However, in addition to their high cost, these techniques are dependent on having specialized equipment. Thus, there is a need for a simple genotyping method that can be implemented in a resource-limited environment.
Methods:
We developed a strategy for SNP genotyping based on T7 Endonuclease I cleavage and an enzyme-linked microparticle immune assay. Using this method, we genotyped two common SNP sites (rs11526468 and rs12979860). The quality of the genotyping process was validated.
Results:
Although a 70% false-negative rate was observed, no false-positive reactions were found. Therefore, multiple parallel repeat reactions can offset the possibility of mutation detection failure.
Discussion:
This method employs a duplicate reagent-dependent procedure, and therefore has the potential for integration into a portable kit for field utilization.
Introduction
Large-scale microarray and next-generation sequencing techniques have made genome-wide single nucleotide polymorphism (SNP) genotyping possible (Shendure et al., 2004; Sobrino et al., 2005; Stefl et al., 2013; Tan et al., 2013). These platforms, such as Solexa (Illumina), GSFLX (Roche), and SOLID (Applied Biosystems), could be used for identifying novel SNPs, which would influence human traits or common diseases (Flint-Garcia et al., 2003; Wang and Liu, 2006). Furthermore, a large variety of techniques for mid-throughput SNP genotyping have also been developed, such as TaqMan (Livak et al., 1995), Amplifluor (Myakishev et al., 2001), and SNP arrays (Matsuzaki et al., 2004; Shen et al., 2005). These methods could provide important information for clinical diagnosis (Nadeau, 2002; Eberle, et al., 2007; Hamano et al., 2010; Huang et al., 2012; Mates et al., 2012; Zhang et al., 2012; Jang et al., 2013). However, in addition to their high cost, these techniques are dependent on specialized equipment, which are not practical for small workstations and a field-based environment. Concerning populations in areas of the world with low technology, or with the emergence of SNP identification in which samples could not transfer to the laboratory, there is a need for a simple and rapid genotyping method that can be implemented in laboratories lacking access to sophisticated equipment or in mobile workstations.
In this article, we developed a simple and cost-effective strategy for SNP genotyping of small populations based on T7 Endonuclease I cleavage and the enzyme-linked microparticle immune assay method. Using the method, we genotyped two common SNP sites (rs11526468 and rs12979860), and the quality of the genotyping process was validated.
Materials and Methods
Ethics statement
The genomic DNA of 64 donors was collected and analyzed by the T7 endonuclease-based method. The information of the donors is described in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/gtmb). The donor genomes were extracted before sequencing PCR by Sangon Biotech (Shanghai, China). The associated protocol was approved by the Research and Ethics Committee of the Fourth Military Medical University. All participants provided written informed consent, in accordance with the Declaration of Helsinki, before collection of blood samples.
Sample preparation and reaction processing
Genomic DNA was extracted using the E.Z.N.A. Blood DNA Kit (Omega Bio-tek, Norcross, GA). The PCR was conducted using a PCR premix (TaKaRa, Mountain View, CA) and Taq enzyme (TaKaRa). The mixtures in different steps were purified using the miroElute DNA Clean Up Kit (Omega Bio-tek). Dynabead MyOne Streptavidin T1 Beads (Thermo Fisher Scientific, Waltham, MA) were used for product isolation, and the T7 Endonuclease I enzyme (New England Biolabs, Ipswich, MA) was used for cleavage of the unpaired sequences. The anti-digoxigenin horseradish peroxidase (HRP) antibody (Abcam, Cambridge, MA) and substrate 3,3′,5,5′-tetramethylbenzidine (TMB) (Beyotime, Shanghai, China) were used for color reaction in the final stage of detection. The genotyping primers were conjugated with biotin, whereas the probes were conjugated with digoxin. All primers and probes (Supplementary Table S3) were ordered from Sangon Biotech.
Results
Individual genome collection and pilot studies
To determine the reliability of this method, we performed a pilot analysis of the samples. Genomes from 64 donors were first analyzed by direct sequencing. Specifically, we examined the genotypes rs11526468 and rs13284404, which are located at the exon of the gene Sema4D. We found that 30 donors were genotype G/G, 25 donors were G/A, and 9 donors were AA in the rs11526468 site. However, all of the rs13284404 genotypes were G/G, which was subsequently used to determine the failed detection levels (Supplementary Table S2).
Assay strategy and method application
The design strategy is summarized in Figure 1. The primer sequences were consistent with the upstream region near the SNP site but did not include the SNP site. To achieve suitable Tm values, the primer lengths were designed to be between 20 and 28 bp. The primers were biotinylated on the 5′-end of the sequence. The SNP-specific probes were complementary to the nearby sequence of the SNP site. The probe sequence comprised three parts: 18- to 20-bp sequence complementary to the region upstream of the SNP site; nucleotide complementary to varying areas of the SNP site; and 10-bp sequence downstream of the SNP site. The probes were tagged with digoxigenin on the 5′-end of the sequence. As the SNP site includes different variants, the probes include sequences that target to specific SNP variants. The negative control probe contains a nucleotide that is not complementary to any of the SNP variants.
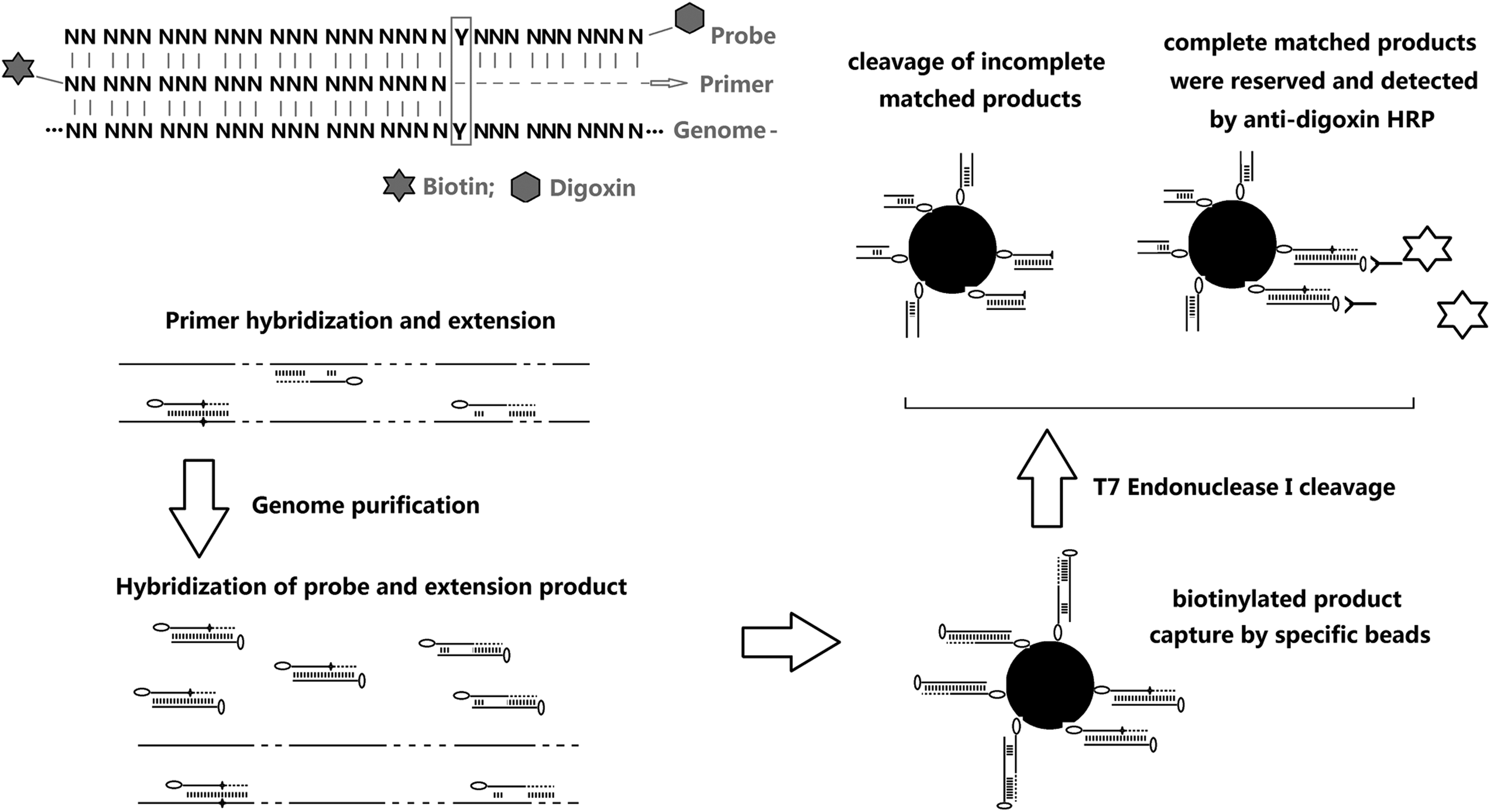
Primer design and genotyping strategy of the SNP determination method. Briefly, after genome extraction, total DNA was hybridized and annealed to each primer. The products were then purified and separately hybridized to each probe. The mixtures were purified, and the hybridization products were isolated from other sequences using streptavidin beads. Finally, sequences without complete complementary binding were cleaved using the T7 Endonuclease I enzyme. The fully complementary sequences were conserved and checked using the HRP-linked immunoassay reaction. HRP, horseradish peroxidase; SNP, single nucleotide polymorphisms.
The polymorphisms of the SEMA4D SNP sites, rs11526468 and rs13284404, were examined as a test of the T7 Endonuclease I method. Briefly, primers and probes were designed (Fig. 2A). Genomic DNA from 64 donors was isolated using the Blood DNA Kit. Extracted genomic DNA (10 μL, 100 ng/μL) was added to each reaction system. Other reaction components included primers (1 μL, 10 μM), Taq polymerase, and premixed buffer. Sequence extension was performed after fully mixing. The samples were incubated at 95°C for 5 min and gradually cooled down to a temperature below 35°C.
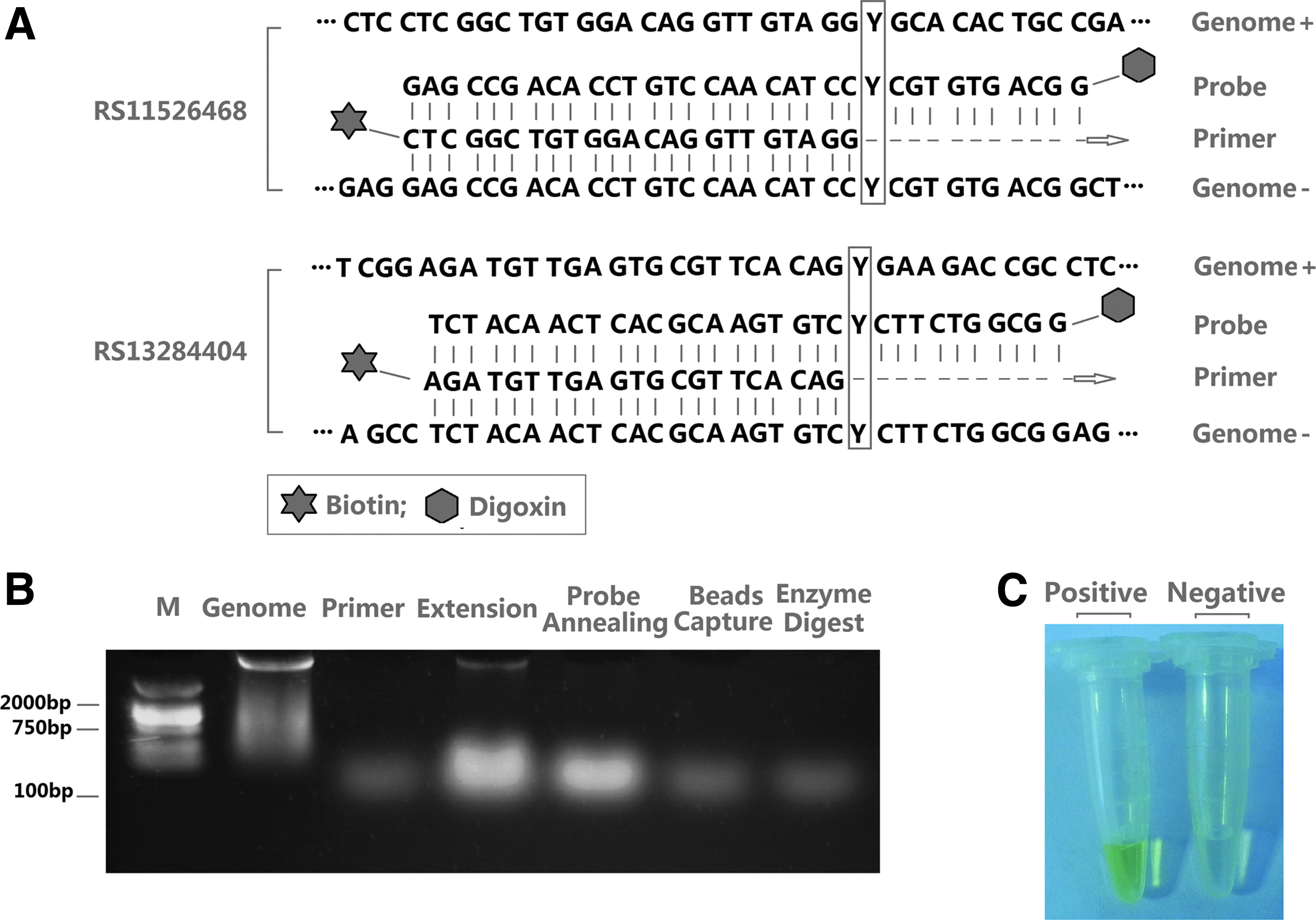
Primer and probe design and SNP analysis of rs11526468 and rs13284404.
The PCR buffer was then removed using a DNA Cleanup Kit, and the purified sample was used for probe hybridization. Each probe (1 μL, 10 μM) was added to the extension product and incubated at 95°C for 5 min, then gradually cooled to a temperature below 35°C. Resuspended streptavidin beads (150 μL) were used to capture the biotin-labeled extension product, and the unpaired sequences were removed by 40 U (4 μL) T7 Endonuclease I cleavage enzyme. The reactions of T7 endonuclease digestion were performed at 25°C for 20 min. Then, anti-digoxigenin antibodies were added to the system and incubated for 10 min. Finally, the sample was washed with phosphate-buffered saline buffer before carrying out the TMB-induced color reaction for observation. The products in each step were validated (Fig. 2B), and the results were confirmed by the detection of positive color (yellow), which confirmed a genotype same as the probe (Fig. 2C).
Genotyping analysis and strategy optimization
Of the 64 participant samples examined in this study, 22 (34.38%) samples showed failed detection for the A reaction of rs11526468 as well as 40 (62.5%) samples for the G reaction of rs11526468. Moreover, for rs13284404, the missed detection rate was 0 for the A reaction and 27 (71.88%) for the G reaction. Therefore, regardless of A or G reaction, all negative samples could be correctly identified as negative, whereas positive ones would result in a missed detection, with a ratio between 64.71% and 72.73% (Supplementary Table S4; Fig. 3B).
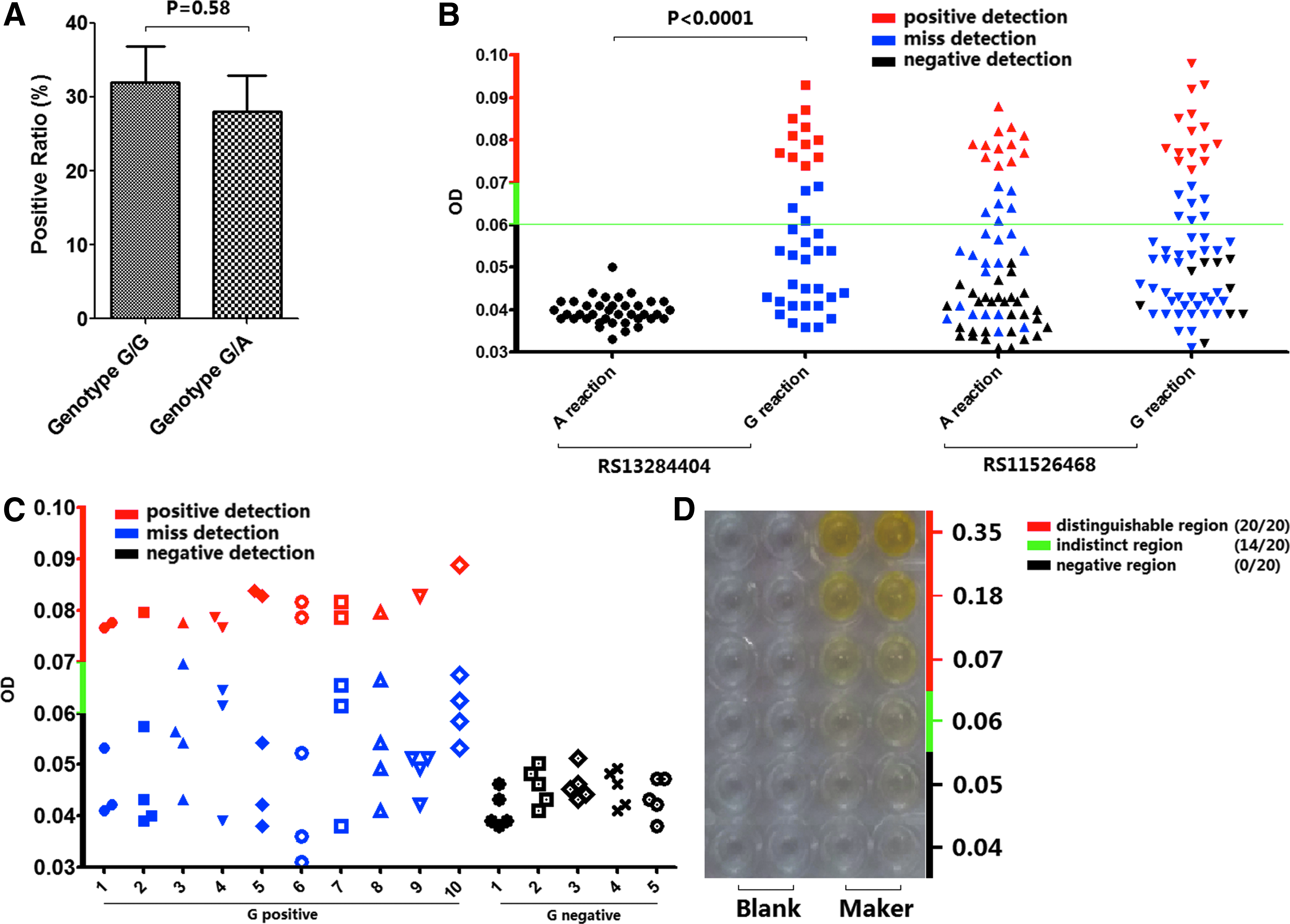
Genotyping analysis of rs11526468 and rs13284404 combined with OD.
To further determine if the missed detection was introduced by the sample specifically or was a random occurrence in the reaction process, we employed 10 samples with G-positive genotype (G/G or G/A) and 5 samples with G-negative genotype (A/A) for repeatability testing. Each sample was tested five times with G reactions at the same condition. Based on these findings, the missed detection ratio should be <80%, which means that five repeats with one sample may provide sufficient accuracy for identification (Fig. 3C). The reaction sensitivity between homozygote (G/G) and heterozygote (G/A) was also examined, and no statistically significant difference was detected (Fig. 3A).
As the color reaction was employed for result validation, we examined the color-distinguishable region associated with the optical density (OD) (450 nm). Twenty volunteers were evaluated for their ability to differentiate the resulting colors. When the OD value was higher than 0.07, all tested individuals could correctly perceive them; in contrast, when the OD was lower than 0.06, a negative conclusion was given by all volunteers (Fig. 3D). As an actual negative sample cannot have an OD value higher than 0.06 (Fig. 3B), false-positive judgment would not have been introduced.
Discussion
Genome exploration through high-throughput SNP genotyping has been rapidly developed and is leading to a deeper understanding of the relationship between genotype and phenotype (Feltus et al., 2004; Hillier et al., 2007). While this approach has improved the efficiency of the genotyping process, it is largely dependent on specialized machinery (Lyamichev et al., 1999; Kolpashchikov, 2006; Xiao Y, et al., 2009). In this study, we developed a method which has potential to estimate SNPs without specific equipment. By genome hybridization and single extension, the target SNP sequence is first enriched. Unlike machine-dependent methods, this novel method involves SNP analysis using a series of reagents for DNA manipulation, which could be integrated into a portable reagents kit for field operation. Other enzyme-based detection schemes had also been developed, while the special materials such as nanoparticles were employed (Wu et al., 2014; Ngo et al., 2016), which require costly modified probes. However, our method involves SNP analysis using a series of reagents for DNA manipulation, which is cheaper and could be detected by naked eye. Moreover, the SNP genotyping approach uses commercially available DNA purification kits and the HRP-link immune reaction system, which makes the method more practicable.
Footnotes
Acknowledgment
This work is supported by grants from National Natural Science Foundation of China (NSFC) 81670529 (C.F.) and 81772167 (X.W.).
Author Disclosure Statement
The authors declare that there are no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.