
Abstract
Objective:
Heterozygous pathogenic variants in the COL2A1 gene result in several clinical features including impaired skeletal growth, ocular and otolaryngological abnormalities. Missense mutations in the triple helical region of the COL2A1 protein have been associated with lethal spondyloepiphyseal dysplasia (SED). In this study, we aimed to identify the underlying cause of a case of SED congenita (SEDC) in a 27-month-old child.
Materials and Methods:
A patient who was diagnosed initially with osteochondrodysplasia underwent a detailed clinical and radiological examination to obtain a conclusive diagnosis. The patient did not show any clinical features of hypochondrogenesis. Whole exome sequencing of the COL2A1 gene was carried out to identify the underlying genetic cause of the disorder.
Results:
Variant annotation and filtration detected a heterozygous missense mutation c.1357G>A (p.G453S) in the exon 21 of the COL2A1 gene of the proband which was confirmed by Sanger sequencing. Neither parent carried the mvariant suggesting this was a new mutation.
Conclusion:
The COL2A1 mutation (c.1357G>A), identified in this case, results in more mild phenotype than other missense mutations in exon 21 which are known to cause lethal hypochondrogenesis. We showed, for the first time, that a missense mutation (p.G453S) in the triple helical region of the alpha 1 (II) chain of the COL2A1 protein underlies SEDC and is not always lethal.
Introduction
Mutations in COL2A1 (collagen type 2 alpha 1) gene is known to cause variety of clinical conditions including spondyloepiphyseal dysplasia congenita (SEDC) (Liu et al., 2016; Sangsin et al., 2016; Xiong et al., 2018), Kniest dysplasia (Al-Hashmi et al., 2013; Barat-Houari et al., 2016b), Stickler syndrome (Higuchi et al., 2017; Huang et al., 2018), osteoarthritis with chondrodysplasia (Jakkula et al., 2005), Legg-Calve-Perthes disease (Miyamoto et al., 2007; Kannu et al., 2011), and other skeletal conditions (Reinartz et al., 2017; Güneş et al., 2018; Liu et al., 2018). Conditions that arise as a result of COL2A1 gene mutations are collectively termed type II collagenopathies (Barat-Houari et al., 2016a). Both autosomal recessive and autosomal dominant phenotype have been reported due to COL2A1 mutations (Tham et al., 2015; Barat-Houari et al., 2016c; Xiong et al., 2018). Clinical characteristics of COL2A1 mutations include skeletal deformities (epiphyseal dysplasia, metaphyseal deformities, disproportionate short stature, midface hypoplasia, spinal deformity, and hypoplastic pelvis), ocular abnormalities, deafness, and cleft palate. However, intrafamilial and interfamilial phenotypic variabilities are common (Nagendran et al., 2012).
SEDC exists, in most cases, as an autosomal dominant condition with skeletal deformities at birth. SEDC is characterized by short-trunk dwarfism, odontoid hypoplasia, cervical spine subluxation, scoliosis, kyphosis, lumbar lordosis, coxa vara, genu valgum, clubfoot, metaphyseal changes, abnormal epiphyses, and flattened vertebral bodies. Extra-skeletal features include midface hypoplasia, sensorineural hearing loss, myopia and/or retinal degeneration with retinal detachment, micrognathia, thoracic hyperkyphosis, and cleft palate (Xia et al., 2007; Mark et al., 2011; Zhang et al., 2011; Veeravagu et al., 2013; Xu et al., 2014; Liu et al., 2016; Higuchi et al., 2017; Xiong et al., 2018).
Here, we studied a patient who was initially diagnosed as having osteochondrodysplasia. Whole exome sequencing identified a heterozygous missense mutation in the COL2A1 gene. Detailed clinical examination followed by extensive genetic analysis confirm the case as SEDC.
Materials and Methods
Patient
A 6-month-old female was referred to the Centre for Genetics and Inherited Diseases (CGID). The affected individual was delivered by a caesarean section. The subject underwent detailed clinical and radiological examination at the department of genetics, Madinah maternity and children hospital (MMCH), Saudi Arabia. The study was approved by Ethical Review Board of MMCH, Almadinah Almunawarah, Saudi Arabia. Signed informed consents were obtained from parents of the affected individual for genetic analysis and publication of photographs. Parents were called to the CGID and interviewed to record history of incidence of the disease and the disease status. Profile photographs of patients were taken for presentation/analysis of facial dysmorphism. Head circumferences were measured to exclude the microcephalic feature in the patient. Plain chest radiographs and an echocardiogram were taken to rule out any heart diseases. A computed tomography (CT) scan of the brain of the affected individual was also performed to rule out any brain abnormalities. Blood and urine biochemical tests were also performed to exclude any metabolic disorders.
Extraction of genomic DNA and targeted sequencing
Peripheral blood samples were collected from both parents and an affected child in an EDTA coated tube. Nucleic acid was extracted using QIAquick DNA extraction kit. DNA was quantified by Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA) and Qubit fluorometer (Thermo Fisher Scientific).
Initially, 25 candidate genes (based on functional and expression data) were sequenced by Sanger approach. Ensembl genome browser * was used to download the genomic sequences of the candidate genes. Primers were designed using primer3 software † to amplify the exonic regions and flanking sites. The patient's genomic sequences were aligned to the reference sequence using BIOEDIT sequence alignment editor version 6.0.7 (Ibis Biosciences, Inc., Carlsbad, CA).
Sequencing of all the protein-coding genes
Genomic DNA from the affected individual was subjected to exome sequencing. Library preparation and exome enrichment was done by using SureSelect Target Enrichment kit (Agilent Technologies, Santa Clara, CA). DNA sequencing and cluster generation was carried out on NextSeq500 machine (Illumina, San Diego, CA). Exome sequencing protocol is described elsewhere (Hashmi et al., 2016). For analysis purposes, a BaseSpace (Illumina) was used to align fastq files to the reference genome. Variants were called by using genome analysis tool kit (GATK). Genomic variants were annotated and filtered by VariantStudio (Illumina).
Results
Clinical and radiological analysis of the patient
The affected individual, who is now 27 months old, was born to phenotypically normal parents and was recruited for this study (Fig. 1). Heights of both parents were in normal range. Father is 36 years old with a height of 165.4 cm and mother is 29 years old and her height is 153 cm.
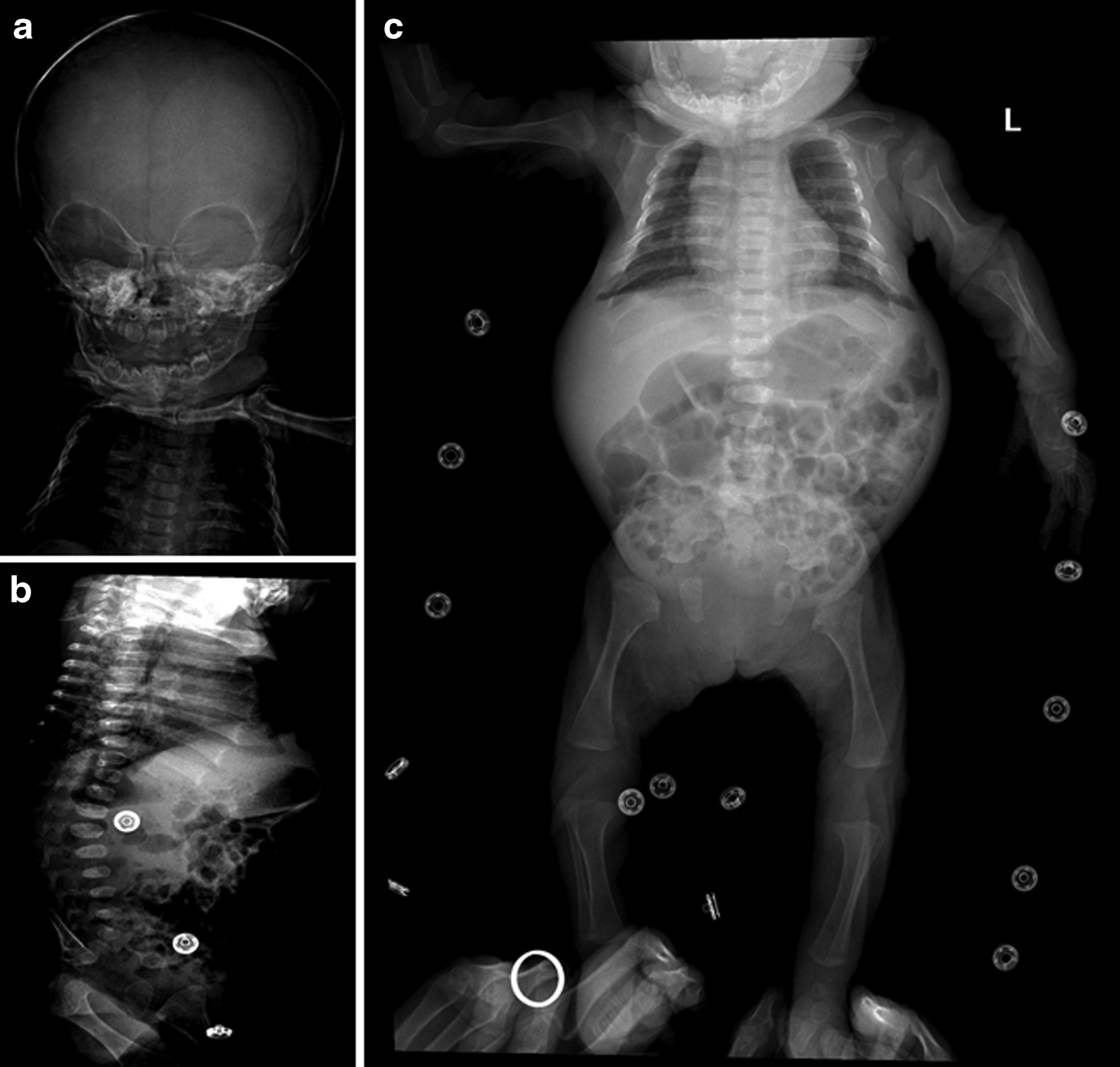
Radiographs of an affected child showing typical features of spondyloepiphyseal dysplasia congenital.
The age of the affected individual was 6 months at the time of recruitment. We were unable to record the growth parameters at the time of birth. However, growth parameters at the age of 6 months were recorded as a height of 39 cm less than 3rd percentile, head circumference (occipitofrontal diameter) of 42 cm on the 75th percentile, and a weight of 5.5 kg corresponding to the 25th percentile. Cognition and mentality were found preserved. A facial dysmorphism was found in terms of relative macrocephaly, flat midface with frontal bossing, hypertelorism, and depressed nasal bridge with a high arched palate. A patient was presented with a cleft of the soft palate. A short neck with severe trunk shortening and a barrel chest (pectus carinatum) deformity was also observed. Patient exhibits features of disproportionate short stature. Involvement of the spine was also observed with kyphoscoliosis and mild lumbar lordosis. A rhizomelic limb shortening with coxa vara; bilateral clubfoot deformity; and a mild degree of restricted movements of large joints including elbow, hip, and knee joints was also observed. Laboratory investigations including measurement of amino acid levels, creatine, and creatinine level were normal, making a metabolic disorder unlikely. Ophthalmic examination excluded issues related to eye/vision. Abdominal ultrasonography and echocardiogram values were in normal range. However, the abdomen was found mildly protuberant without any organomegaly. CT scanning of brain showed periventricular white matter hypo-density prominent in the frontal lobes suggestive of early hypoxic ischemic changes.
Skeletal survey showed relatively large calvarium, bell-shaped chest, platyspondyly, posterior wedging of the vertebrae, long bones metaphysical flaring, and absent pubic bones suggestive of SEDC (Fig. 1).
Screening candidate genes did not reveal any pathogenic mutation
Patient was referred to CGID with multiple clinical features including asphyxiating thoracic dystrophy, hypophosphatasia (based on low alkaline phosphatase level), atelogenesis defect, mild facial hypoplasia, short thoracic cage with short extremities, osteochondrodyspasia, short stature, and cleft palate. For this reason, we first selected and screened 25 previously known candidates genes that cause abovementioned phenotypes. This includes ALPL, CBFA1, CEP120, DYNC2H1, DYNC2L11, FLNA, FLNB, IFT10, IFT140, IFT172, IFT52, IFT80, KIAA0586, NEK1, RUNX2, RUNX3, SLC26A2, SOX9, TCTEX1D2, TTC21B, WDR19, WDR19, WDR34, WDR35, and WDR60. Sequencing data analysis identified an 18-bps heterozygous in-frame deletion variant (c.243-260delGGCGGCTGCGGCGGCGGC) in the RUNX2 gene. Initially, we considered this variant as a causal mutation for the patient's phenotype based on previous report (Zeng et al., 2018). However, further analysis of variant revealed that it is present in high frequency in variety of genome variation databases. Moreover, segregation analysis discovered the presence of variant in mother as well. Therefore, this variant was considered as a population polymorphism (Hashmi et al., 2018).
Exome data analysis identified a novel mutation in COL2A1 gene
WES libraries were generated using 51 Mb SureSelect V6 kits (Agilent Technologies). A 103 bps paired end reads were obtained with an average read depth of 130. Reads were aligned to the human genome reference sequence hg19 using Burrows-Wheeler Aligner (BWA). In total, more than 97,000 variants were obtained. Different filters were applied to obtain candidate variants in the coding regions and exon-intron splice junctions. The details of filtering steps are described elsewhere (Alkhiary et al., 2017). An autosomal recessive model did not reveal any variant of interest. A de novo model was used to select heterozygous alterations with minor allele frequency of <0.0l, and predicted protein effect of “damaging” identified a novel heterozygous missense variant in COL2A1 gene (ENST380518: c.1357G>A; p.Gly453Ser). The detected variant causes the substitution of a glycine into a serine (hydrophobic to hydrophilic substitution) residue at position 453. It is absent in gnomAD and dbSNP databases. Multiple in silico tools predict a deleterious effect of this missense change.
Sanger sequencing validate the presence of variant
Reference sequence of COL2A1 (ENST00000380518.7) was obtained from the ensembl genome browser. ‡ A primer pair (forward: 5′-GACACCCCTCCCTTCTTCTC-3′ and reverse: 5′-ACACCAGATTCTCTCCAGGG-3′) for polymerase chain reaction amplification of the candidate variant was designed using Primer3Plus software. ** The exonic segment was amplified in the DNA of patient and both parents followed by their sequencing using BigDye Terminator v3.1 Cycle Sequencing Kit and ABI 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA). Sequence variant was identified through BioEdit sequence alignment editor version 6.0.7. ‡‡ Patient DNA sequence showed missense variant (c.1357G>A) in heterozygous state, however, parental DNA analysis and DNA analysis of unaffected population controls did not have the variant (Fig. 2).

Sequencing chromatogram showing partial sequence of exon 21 of the COL2A1 gene.
Discussion
Type II collagen is an extracellular protein of cartilaginous tissue and is required for normal endochondral development and cartilage homeostasis. NAD-dependent protein deacetylase Sirtuin 1 (SirT1) binds to the chromatin regions of COL2A1 promoter and enhancer and thus enhances COL2A1 gene expression (Dvir‐Ginzberg et al., 2008). Moreover, it has been shown that Set7/9 forms a protein complex with SirT1 on the promoter region of COL2A1 and enhances COL2A1 expression (Oppenheimer et al., 2014).
In this study, whole exome sequencing was performed on a DNA sample from a patient presented with osteochondrodysplasia. Data analysis identified an 18-bps heterozygous in-frame deletion variant (c.243-260delGGCGGCTGCGGCGGCGGC) in the RUNX2 gene. Initially, we considered this variant as a causation mutation for the patient's phenotype based on previous report (Zeng et al., 2018). However, further analysis of variant revealed that it is present in high frequency in different databases, apparently healthy individuals and in the DNA of healthy mother (Hashmi et al., 2018). Three-dimensional structures of the wild-type and mutant RUNX2 protein (p.Ala82_Ala87del) were analyzed and it was found that both wild-type and mutant proteins show similar secondary structure patterns. This lead to the argument that the 18-bps heterozygous in-frame deletion variant in RUNX2 gene is a population polymorphism and not a pathogenic mutation (Hashmi et al., 2018). Further filtration of exome variants detected a heterozygous missense mutation (c.1357G>A) in the COL2A1 gene. This variant substitutes a bulky serine residue for a glycine amino acid (p.G453S) in a triple helical region of alpha 1 (II) chain of the COL2A1 protein. Flanking region of this variant in exon 21 was amplified in DNA samples from proband and both parents to validate the presence of variant. Sanger sequencing validate the presence of variant in proband, however, it is absent in the DNA samples of both parents.
Heterozygous missense mutations in the triple helical region of alpha 1 (II) chain of the COL2A1 protein cause dominant negative effects. These mutations normally substitute bulky amino acid for glycine residues and thus may result in the assembly of abnormal alpha 1 (II) chain into trimeric collagen molecules. Abnormal collagen molecules cause spectrum of SED (Nishimura et al., 2005). Specifically, mutations in the exon 21 and 22 lead to the clinical phenotype of hypochondrogenesis, a lethal SED. Most carriers of heterozygous missense mutations in exon 21 and 22 died in the first year of life (Nishimura et al., 2005). However, in this case, the patient showed typical features of SEDC and the patient is alive at the age of 2.5 years. Characteristics features of hypochondrogenesis including mesomelia, square-shaped short iliac bone, short and broad neck of the femur, elongated distal fibula, short pedicle of lumbar spine, dorsal concavity of lumbar vertebral body, and shortening of distal ulna have not been observed in this case. Phenotypic variability can be attributed to incomplete penetrance or variable expressivity. Multiple factors have been identified as an underlying cause of reduced penetrance and variable expressivity including genetic modifiers, allelic variations, and complex genetic and environmental interaction (Cooper et al., 2013). We hypothesize that mutation(s) in regulatory regions are responsible for phenotypic variability and reduced penetrance. Mutations in regulatory regions cannot be detected using whole exome sequencing approach and therefore, the whole genome sequencing may reveal the deep intronic or regulatory regions variants as an underlying cause of the phenotypic variability and reduced penetrance in this patient.
In conclusion, the missense mutation (c.1357G>A; p.G453S) in the triple helical region of alpha 1 (II) chain of the COL2A1 protein underlies non-lethal SEDC.
Ethics Approval and Consent to Participate
All laboratory experimentations were approved by the Ethical Review Committee of Taibah University. All participants signed informed written consent.
Footnotes
Authors' Contributions
A.A. performed Sanger validation; F.A. recruited the case and performed clinical investigations; Y.N.K. performed skeletal survey and drafted X-ray report; S.A., J.A.H., and A.M.A. performed exome sequencing; A.U. performed exome data analysis; S.B. designed the study and prioritized variants; J.A.H. wrote the initial draft of the article. All authors contributed to and have approved the final article.
Acknowledgments
We are grateful to all the participants for their willingness to participate in this study. We are also thankful to Dr. Amir Ali Khan who performed thorough English editing and corrected the grammatical mistakes in the article.
Author Disclosure Statement
No competing financial interests exist.