
Abstract
Purpose:
Congenital aniridia is a kind of panocular disorder characterized by the absence of the iris in both eyes. Paired box 6 (PAX6) gene mutations have been identified as the most common cause of congenital aniridia. The aim of this study was to reveal the genetic defect in PAX6 in a Chinese family with congenital aniridia.
Methods:
Twelve individuals from a three-generation Chinese family were recruited. All the family members underwent comprehensive ophthalmologic examinations. The entire coding region of PAX6 was amplified by polymerase chain reaction, followed by direct Sanger sequencing. Possible structural and functional changes of protein were predicted by bioinformatic analysis using SIFT and Polyohen-2.
Results:
Among all the 12 members, four were clinically diagnosed with congenital aniridia. A novel heterozygous mutation c.275G>A (p.R92Q) in exon 6 of PAX6 was identified in all the patients, but not in the unaffected individuals or 1186 healthy subjects. This missense mutation is a G-A transition, converting Arginine (R) to Glutamine (Q) at amino acid 92. The substitution of amino acid in the PAX6 protein changed the local charge density and was predicted to damage the normal protein function.
Conclusions:
Our study identified a novel mutation of PAX6 responsible for congenital aniridia in a Chinese family, which may contribute to understanding the molecular basis and clinical diagnosis of congenital aniridia.
Introduction
Congenital aniridia (OMIM: 106210) is a rare, bilateral, and panocular abnormality characterized by partial or complete absence of the iris (Nelson et al., 1984). The incidence is ranging from 1:64,000 to 1:96,000 for different ethnic groups. Almost two-thirds of the patients are autosomal dominantly inherited.
In addition to the variable iris hypoplasia, congenital aniridia is usually accompanied with other eye anomalies such as lens opacity or dislocation, nystagmus, glaucoma, aniridia-related keratopathy (ARK) caused by corneal limbal stem cell deficiency, foveal or optic nerve hypoplasia with obvious compromised visual acuity (Mackman et al., 1979; Hingorani et al., 2009). Aniridia is also associated with some systemic disorders, including Wilms tumor aniridia genital anomalies retardation syndrome, a kind of rare genetic condition composed by Wilms tumor (a rare form of kidney cancer), aniridia, abnormalities of the genitalia and urinary tract, and intellectual disability (Fischbach et al., 2005).
Usually, clinical diagnosis of congenital aniridia is further confirmed by the identification of Paired box 6 (PAX6) gene mutations (OMIM: 607108) (Brown et al., 1998; Hu et al., 2015). The PAX6 gene is a member of the paired box gene family, spanning about 22 kb at chromosome 11p13. It encodes a transcription regulator that includes two highly conserved DNA-binding domains (a paired box and a paired type homeobox), and contains 14 exons with 3 isoforms of transcripts by alternative splicing (Chalepakis et al., 1993). This gene plays an essential role in eye development, as well as morphogenesis of other organs (Kozmik, 2008; Yan et al., 2011). As an highly conserved protein of 436 amino acids, the metazoan-bearing PAX6 protein consists of three domains: a proline/serine/threonine-rich domain in the C-terminus, a paired domain (PD) in the N-terminus, and a homeodomain between them (Ton et al., 1991).
Mutations in PAX6 are responsible for various severe ocular disorders, such as aniridia, cataract, glaucoma, and myopia (Jordan et al., 1992; Glaser et al., 1994; Ng et al., 2009), all of which could eventually be the cause of blindness. In 1992, Jordan first demonstrated the crucial role of PAX6 gene for normal human eye development by reporting deleted or mutated PAX6 gene in aniridia patients (Jordan et al., 1992). To date, at least 800 mutations have been submitted in the PAX6 mutation database, and among these more than 300 mutations were described in aniridia patients (human gene mutation database; Cvekl and Callaerts, 2016). In this study, we used gene sequencing technology to reveal the genetic defect in PAX6 in a Han Chinese family with congenital aniridia.
Materials and Methods
Subjects
A three-consecutive-generation Chinese family with congenital aniridia, totally 12 individuals, was recruited from Shandong Provincial Hospital affiliated to Shandong University (Fig. 1). Our study was performed in accordance with the tenets of the Declaration of Helsinki and approved by the Institutional Review Boards of Sichuan Provincial People's Hospital. Written informed consent was signed by each participant before the study. The 792 unrelated healthy subjects from Shandong Provincial Hospital and 394 unrelated healthy subjects from Sichuan Provincial People's Hospital were recruited as controls when they attended the eye clinic for annual examinations.
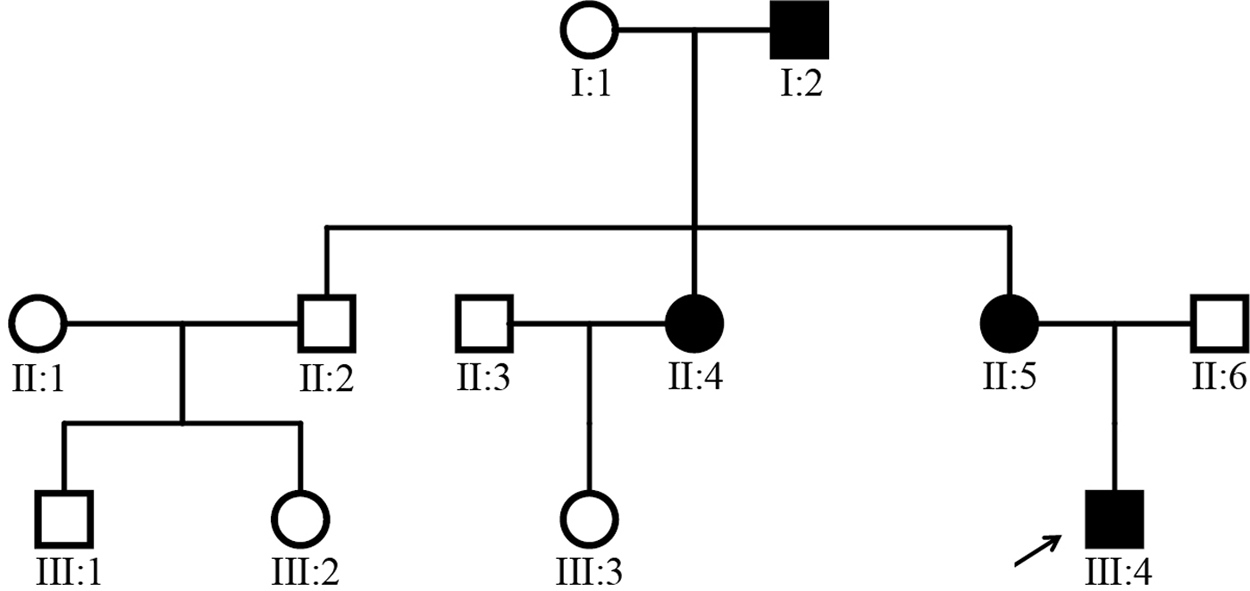
Pedigree of the family with congenital aniridia. Solid symbols indicate affected individuals. Open symbols indicate unaffected individuals, and arrow indicates the proband of this family.
Clinical diagnosis
Four of the 12 family members were diagnosed with congenital aniridia, and non-consanguineous marriages were found in the family. Clinical information of all affected family members was summarized in Table 1. Each member in this family underwent general clinical evaluation and thorough ocular examination: visual acuity assessment, anterior segment examination, intraocular pressure measurement, fundus examination, and orthoptic evaluation, as well as the examination of physical malformations and neurological deficits. The patients with aniridia should meet the inclusion criteria that partial or complete absence of the iris was detected before 18 years of age at the time of diagnosis. All of the 1186 normal control subjects were Han Chinese without any related phenotypes and family history of ophthalmic disease. Individuals were also excluded if they had any of the abnormalities, including any vascular defects, central neural system diseases, ocular disorders, and skeletal deformities.
Summary of Clinical Manifestations for Patients in the Family
ARK, aniridia-related keratopathy; BCVA, best corrected visual acuity; L, left eye; PCO, posterior capsule opacification; R, right eye.
Mutation screening and bioinformatics analysis
Genomic DNA were extracted from peripheral venous blood samples. The coding sequences of PAX6 (NM_000280.4, transcript variant 1) were amplified by polymerase chain reaction using a MyCycler thermocycler (Bio-Rad, Hercules, CA). Sequencing primers of each exon were designed using Primer 5.0 (Supplementary Table S1). To check for the sequence conservation of the residues, we conducted multiple sequence alignment of the human PAX6 protein along with other PAX6 proteins across different species.
Results
Clinical findings
The inheritance pattern of this family was indicated in the familial pedigree (Fig. 1). In this study, we did not detect any physical malformations or neurological deficits among the patients in this family. All the four patients (I:2, II:4, II:5, and III:4) presented severe visual impairment and glare in both eyes since their early childhood. They all suffered from total absence of bilateral irises and reading difficulties, and showed similar clinical symptoms, including low visual acuity, complete aniridia, cataract, significant photophobia, and horizontal nystagmus (Table 1 and Fig. 2). Gonioscopic evaluation of the angle presented no alterations, and intraocular pressure was normal in all affected members. Examination of anterior segment of the proband (III:4; age 12 years) and his mother (II:5; age 35 years) showed scattered punctate linear cortex opacities and dense subcapsular central opacity of the lens. Even with the presence of cataract, physiological papillary excavation still could be detected during fundus inspection. Marked foveal hypoplasia was exhibited in optical coherence tomography. The proband also had ptosis in both eyes, as well as exotropia of 30 prism diopters either near or at distance. Upper eyelid margin of the boy rested about 1 mm below pupils. He presented special appearance of raising his eyebrows by contracting the frontalis muscles in an attempt to lift up his dropping upper eyelids. Patient II:4 (age 37 years) and patient I:2 (age 64 years) underwent bilateral cataract surgery 30 and 31 years ago, respectively. Extensive posterior capsule opacification (PCO) occurred in both eyes after surgery. For patient II:4, fibrous and Elschnig pearls mixed PCO took place in her left eye, as well as pearl form of PCO occurred in the right. ARK was not noticed in all the family members except for patient I:2 who had mild achlys and biocular corneal pannus from limbal region in the superior part of the peripheral cornea.
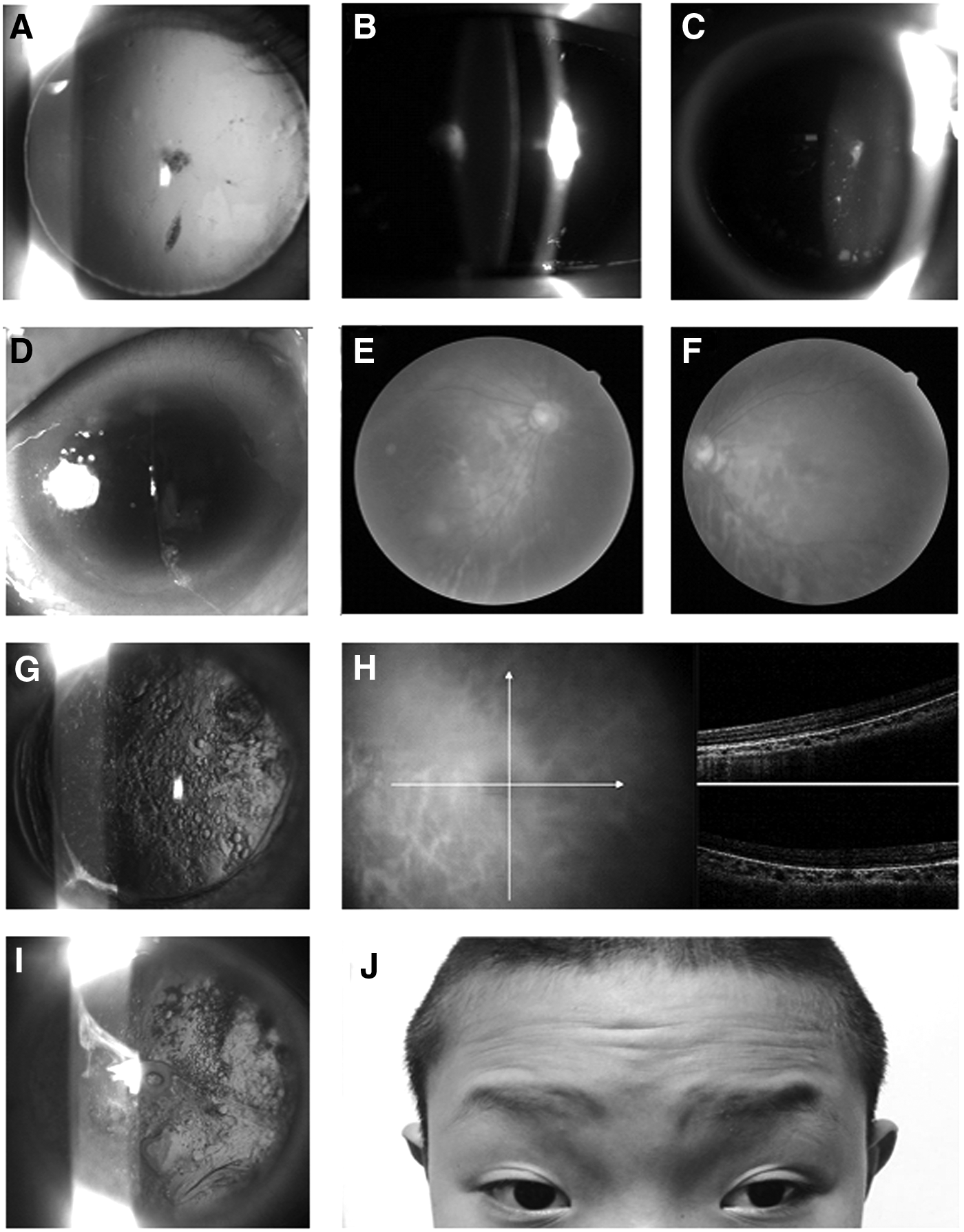
Representative photos of the patients in the family with aniridia.
Mutation screening of PAX6
To detect the genetic defects of patients from this family, we screened the exons of PAX6 by direct Sanger sequencing. It revealed a novel heterozygous mutation c.275G>A (p.R92Q) located at nucleotide 275 in coding sequence of exon 6 from the sequencing analysis of the PAX6 gene (Fig. 3). All the four affected patients (I:2, II:4, II:5, and III:4) presented this missense mutation, while it was not found in other members of the family nor in 1186 healthy control subjects. Therefore, this mutation was co-segregated with the phenotype in the Chinese family recruited in our study. It demonstrated that this novel mutation appeared at highly conserved positions of other PAX6 proteins across different species by comparative amino acid sequence alignment assay (Fig. 3). This heterozygous mutation is a G-A transition, converting Arginine (R) to Glutamine (Q) at amino acid 92 (p.R92Q). This amino acid substitution in the PAX6 protein changed the local charge density and was presumed to be damaging by SIFT and Polyohen-2.

Representative chromatogram of the PAX6 sequence.
Discussion
The PAX6 gene has been known to be a candidate gene for aniridia from 1991 (Ton et al., 1991). Many previous studies confirmed that PAX6 gene mutations were the primary cause of various severe aniridia phenotypes in different ethnic groups. Aniridia-associated PAX6 mutations were detected in 55% of Chinese (Zhang et al., 2011a), 60% of Caucasian (Redeker et al., 2008), 30% of Mexican (Villarroel et al., 2008), 56% of Indian (Neethirajan et al., 2006), 67% of Thai (Atchaneeyasakul et al., 2006), 38-58% of German (Wolf et al., 1998; Zumkeller et al., 2003), 50% of Japanese (Kondo-Saitoh et al., 2000), 79% of Danish (Gronskov et al., 2001), and 83-94% of British (Axton et al., 1997; Robinson et al., 2008) aniridia patients.
The PAX6 gene is located in chromosome 11, and the encoded protein can attach to specific areas of DNA, then initiate and regulate transcription of its downstream gene during embryogenesis (Tzoulaki et al., 2005; Kokotas and Petersen, 2010). Two DNA-binding domains of the encoded protein, the paired domain and the homeodomain, are separated by a linker segment and followed by a C-terminal region, which is abundant in proline, serine, and threonine. Mutations in PAX6 gene or in the enhancer regions can lead to various ocular phenotypes (Glaser et al., 1992).
In the present study, we described a novel c.275G>A (p.R92Q), heterozygous mutation of the PAX6 gene in a Chinese family with congenital aniridia. All the four patients (I:2, II:4, II:5, and III:4) in this pedigree were found to harbor the novel mutation. This mutation was not detected in other family members or in 1186 normal controls. Among public databases, this variant was only found to exist in ExAC database with an allele frequency of 0.0004622 in East Asians (1 in 1000 East Asian individuals) without any clinical features, and was not present in other ethnicities including Africans, Europeans, Latinos, South Asians, and other populations. Taken together, this mutation may be a rare variant at least in East Asians and is likely to be involved in the pathogenesis of congenital aniridia in this pedigree. For aniridia, different kinds of PAX6 mutations were corresponding to variable clinical manifestations. The phenotype/genotype correlation has been established in previous studies (Zhang et al., 2011b; Lee et al., 2014; Chang et al., 2015; Dubey et al., 2015; Yokoi et al., 2016). Although missense mutations were generally associated with mild phenotypes with better visual acuity, partial iris aplasia, and lower rates of foveal hypoplasia (Hingorani et al., 2009), the proband (III:4), his mother, aunt, and grandfather in this family all displayed complete aniridia and low visual acuity, as well as cataract, absence of foveal reflex, photophobia, and nystagmus. None of them showed any evidence of glaucoma, and only patient I:2 presented local abnormal corneal blood vessels.
For the p.R92Q mutation identified in this study, Arginine was replaced by Glutamine in exon 6. Arginine is a charged amino acid, while Glutamine is non-charged. Therefore, local charge density of the PAX6 protein might be changed due to the p.R92Q missense mutation in the NH2 region, which could deteriorate the function of PD of the protein thereafter. As predicted by Pfam and RaptorX, the p.R92Q mutation mainly takes place within the PAX domain, which is an amino acid motif with DNA-binding ability and exerts multiple functions during embryogenesis. This mutation was also located in the helix area of PAX domain, suggesting that it might change the helix structure and affect the binding of the DNA ligand with PAX6. Sequence analysis also indicated that PAX domain was highly conserved among different species and plays an important role in the formation of the eye. Additionally, the mutation is predicted to impair the protein function by Polyohen-2 and SIFT, which implied a possible pathogenic effect of the mutation. Previous studies have demonstrated that most of the PAX6 mutations could lead to a premature termination codon (PTC) in the PAX6 open reading frame among 77% of aniridia cases, whereas only 11.7% of aniridia-associated mutations were missense mutations (Tzoulaki et al., 2005). The PTC-containing mRNA of these patients are degraded by nonsense-mediated decay (NMD) before producing a large amount of truncated proteins (Wen and Brogna, 2008). However, our findings in this study identified a point mutation in aniridia patients, which did not lead to a premature termination. Considering these facts, we could not assume that this mutation leads to the disruption of PAX6 gene expression or affects the transcription of a functional mRNA by NMD. The exact roles of PAX6 in the physiology of embryonic development and pathology of aniridia remain to be fully understood. Further functional analyses are mandatory to better elucidate the actions of PAX6 and the underlying mechanisms of the disease.
In conclusion, a novel heterozygous mutation c.275G>A (p.R92Q) in the PAX6 gene was identified in a Han Chinese family with congenital aniridia in our study. This finding not only expands the mutation spectrum of PAX6-triggered congenital aniridia, but also increases our understanding of its genetic etiology. In addition, it may potentially facilitate genetic counseling and attribute specific treatment of patients with congenital aniridia as well.
Footnotes
Acknowledgments
This study was supported by grants from the Natural Science Foundation of China (81670853); CAS “Light of West China” Program (to B.G.); The Department of Science and Technology of Sichuan Province, China (2019JDJQ0031 and 2016JY0082); Foundation for Technology and Science and Technology Bureau of Chengdu (2018-YF05-00348-SN); project of medical and health technology development program in Shandong province (2017WSB01063). We thank all the participants in this study.
Authors' Contributions
B.G. and Q.W. conceived and designed the study; Y.X., X.L., and L.L. recruited the participants; X.G. and C.Y. performed the experiments; X.L. and B.G. analyzed the data; B.G. wrote the initial draft; Q.W. edited the article for language. All authors critically revised, reviewed, and gave final approval of this article.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.