
Abstract
Background and Purpose:
Mucopolysaccharidosis 1 (MPS1) is an autosomal recessive disorder of a lysosomal enzyme called alpha-l-iduronidase caused by mutations in the IDUA gene. This enzyme is responsible for the degradation of the mucopolysaccharides, heparan sulfate, and dermatan sulfate. Based on clinical features and enzyme deficiency, MPS1 is divided into three subtypes, including a severe subtype (Hurler syndrome), an intermediate subtype (Hurler-Scheie syndrome), and an attenuated subtype (Scheie syndrome). The objective of this study was to characterize the mutation profiles of 17 Iranian patients with MPS1 and characterize the clinical features associated with their genotypes.
Materials and Methods:
Polymerase chain reaction-based sequencing of the IDUA gene was carried out for 10 patients with clinical diagnoses of MPS1 and 50 healthy controls. To estimate the impact of newly identified variants on the structure and function of the encoded alpha-l-iduronidase, in silico analyses was performed.
Results:
Eight genetic variations were detected, including five missense mutations (p.M1L, p.G51D, p.G134V, p.S157P, p.D301E), two nonsense mutations (p.W402* and p.Y343*), and one deletion (p.GFLNYY197-202), among which p.G134V, p.S157P, p.D301E, and p.GFLNYY197-202 were novel variations that had not been previously reported.
Conclusion:
After combining the results of the two previous IDUA gene studies performed on Iranian MPS1 patients and the results obtained from the current study, it is inferred that despite the presence of a number of previously known mutations, about half of the detected variations were unique in Iranian patients.
Introduction
Mucopolysaccharidosis 1 (MPS1) is a lysosomal storage disorder, which occurs as a result of a deficiency in a lysosomal enzyme known as alpha-l-iduronidase (Mckusick et al., 1972; Hopwood and Morris, 1990). Deficiency of this enzyme results in incomplete degradation of heparan sulfate and dermatan sulfate, which accumulate within the tissues resulting in progressive multiorgan involvements (Muenzer, 2011). Severity of the disorder ranges from the most severe form, (Hurler syndrome) (OMIM#607914), to the attenuated form (Scheie syndrome) (OMIM#607016) (Beesley et al., 2001). The clinical manifestations include severe cognitive dysfunction (especially in Hurler syndrome), skeletal malformation, enlargement of spleen and liver, limited joint mobility, restrictive ventilator defects, and upper airway obstruction (Neufeld and Muenzer, 2001; Beck et al., 2014). The IDUA gene is composed of 14 exons with an approximate length of 19 kb. The gene transcript is 2.3 kb in length and produces a 653-amino-acid-peptide (Scott et al., 1990, 1991, 1992). The deficiency or absence of enzyme activity in leukocytes or fibroblasts, and the presence of elevated dermatan sulfate and heparan sulfate in urine are both used as diagnostic tests to confirm the disease.
Two standard treatments for MPS1 are hematopoietic stem cell transplantation (HSCT) and enzyme replacement therapy (ERT). In ERT, human recombinant IDUA (laronidase) is usually administered intravenously every week to ameliorate physical symptoms of MPS1 disease caused by underlying deficit enzyme (Jameson et al., 2013). However, as laronidase is unable to pass through the brain-blood barrier, it is not an effective treatment in the severe form of disease where progressive intellectual impairment is exhibited. HSCT is a better treatment option for patients with the severe type of MPS1 (Hurler syndrome) and can moderate neurocognitive deterioration and significantly improve the functions of various peripheral organs, especially if it is performed during the early stages of the disease.
Variety of mutations in the locus of IDUA gene have been reported, including missense and nonsense mutations, small deletions or insertions, and splice-site mutations (Boelens et al., 2013). These variations are distributed throughout the IDUA gene and, most are confined to a single individual or family, with a few of them occurring commonly in the world population. However, high frequency of specific mutations has been reported in certain geographic regions. Notably, the most common mutations found in Caucasian are W402* and Q70*, with the former (W402*) being present in about half of the mutant alleles in European countries, America, and Australia, and the latter (Q70*) being reported to be noticeably frequent in Scandinavia and Eastern Europe. In comparison, the most frequent variations specific to East Asian populations were c.1190-1G>A, p.Leu346Arg, and c.613_617dupTGCTC/704ins5 (Yamagishi et al., 1996; Lee et al., 2004; Sun et al., 2011; Wang et al., 2012; Kwak et al., 2016).
The appearance of the two nonsense mutant alleles in the gene locus in homozygous or compound heterozygous state is associated with severe phenotype, while existence of at least one missense or splice-site variation leads to a milder form of the disease. DNA polymorphisms have also been identified in the locus of IDUA gene in nonaffected individuals that could probably modify IDUA gene expression and it is also likely that the existence of these polymorphisms with known MPS1 mutations in the gene locus affect severity of MPS1 disorder (Li et al., 2002; Terlato and Cox, 2003).
Our work aimed to identify pathogenic sequence variations in IDUA gene, which cause the development of mucopolysaccharidosis type 1 in Iranian patients, to evaluate a genotype-phenotype correlation and to compare the spectrum of IDUA mutations observed in MPS1 patients from Iranian population to those found in MPS1 patients from other populations.
Materials and Methods
Subjects
Ten unrelated patients with MPS1, including two females and eight males, between 4 and 12 years of age enrolled in our study at Special Medical Center (SMC), Tehran, Iran between October 2014 and June 2017. For the investigation of the mutation spectrum, we also utilized existing data from literature on Iranian patients studied before (Nasr-Esfahani et al., 2007; Yassaee et al., 2017). Before the study, the activity of alpha-l-iduronidase was measured by fluorometric assay utilizing 4-methylumbelliferyl-α-L-iduronide as a substrate. All patients who underwent enzyme activity testing for definite diagnosis showed very low or no detectable enzyme activity. After research subjects signed the informed consent letters, blood was taken from 10 MPS1 patients and the 50 healthy subjects.
Genotyping
DNA genomic extraction from whole blood was performed by the QIAamp DNA Mini Kit (Qiagen GmbH, Hilden, Germany) under the manufacturer's instructions. Subsequently, nine sets of primers for polymerase chain reaction (PCR)-Sequencing were designed with the help of primer 3 software, covering all 14 exons and the flanking intronic sequence of IDUA gene (NCBI Reference Sequence: NG_008103.1). Genomic DNA (100 ng) was amplified in a reaction volume of 25 μL consisting of 1 × NH4 buffer, 2 Mm dNTPs, 0.4 Mm of each primer, and 5 μL Q solution. Subsequently, 5 μL of PCR products were loaded on 1.5% agarose gel with 100 bp marker and visualized under UV light. The resultant products of PCR amplification were eight fragments of different sizes, including exons 1, 2, 3-4, 5-6, 7-8, 9-10, 11, 12, 13-14. Thermal cycling conditions and primer pairs designed for PCR amplification and sequencing of the IDUA gene are shown in Table 1. Amplicons were sequenced on ABI 3100 and Analysis data software finch TV was applied to visualize the IDUA gene sequence of patients. In addition, IDUA sequences of patients obtained from sequencing were compared with a reference sequence on the NCBI website and alterations in the coding region of the gene and exon-intron boundaries were detected.
Forward and Reverse Primer Sequences, Expected Amplicon Sizes, Annealing Temperature, and Cycling Conditions Used for Polymerase Chain Reaction
PCR, polymerase chain reaction.
Bioinformatics analysis
We browsed through mutations listed on online databases HGMD, dbSNA, ExAC, and 1000 genome browser to search for these newly identified variations.
In silico prediction programs, including SIFT, PolyPhen2, mutpred, and mutationTaster, were all employed to demonstrate potential effects of amino acid changes on protein function. The conservation status of Alpha-L-iduronidase-mutated amino acid residues from human were aligned and compared with the respective residues in different organisms using Clustal Omega and T-coffee.
Results
The mutation analysis was successfully completed in a total of 10 patients, including 8 males and 2 females. Screening of all 14 exons and their flanking introns performed by direct sequencing of PCR products resulted in recognition of eight sequence variations (Table 2), comprising four already known mutations (p.W402*, p.M1L, p.Y343*, and p.G51D), three novel missense variations (p.D301E, p.G134V, p.S157P), and one novel deletion (GFLNYY197-202) (Fig. 1). Of the 10 patients examined, one mutant allele was not detectable in patient VII. All of the affected children, except for patient VIII were born to a consanguineous marriage. Surprisingly, patient VIII, who was born to unrelated parents, was homozygous for a missense alteration (p.M1L). This phenomenon, although uncommon, is attributed to high inbreeding in the region, where the parents were originally from, suggesting that intracommunity marriage is a common practice that increases the chances of offspring to be homozygous. There were two independent reports describing seven patients with MPS1 in the Iranian population. The results from these two previous reports combined with the findings in the present study revealed 12 types of genetic pathogenic variations in 17 patients with MPS1 (Fig. 2), including eight missense variations (p.M1L, p.G51D, p.L132R, p.Gly134Val, p.157P, p.D301E, p.W175R, p.P533R), two nonsense mutations (W402*, Y343*), one duplication c.612_615dup (p.S206Lfs*194), and one deletion c.590-17_607del35 (GFLNYY197-202). Overall 33 mutant alleles were found, of which p.W402* was found to be the most common mutation with a frequency of 17.6% (6/34). Two variations (p.L132R and p.M1L) accounted for 11.7% (4/34) each and p.G51D accounted for 8.8% (3/34). The remaining seven variations were only observed in a single individual with a frequency of 5.8% each (Table 2).
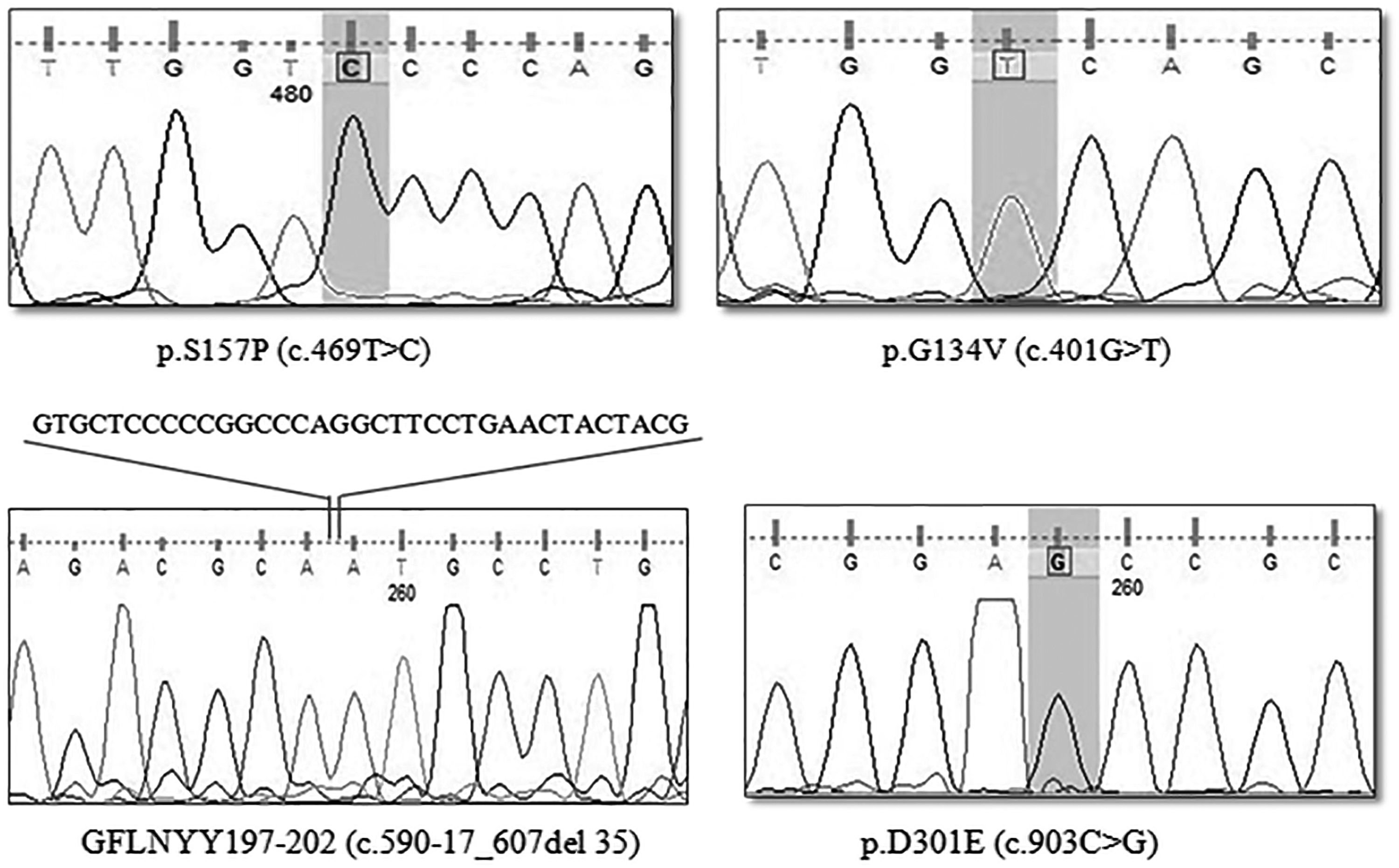
The DNA Sequencing chromatogram displays four novel mutations D301E, S157P, p.G134V, and c.590-17_607del35 detected in patients with Mucopolysaccharidosis type 1.

A schematic of the IDUA gene showing the location of mutations found in Iranian patients.
The Sequence Variations and Their Allelic Frequencies Found in IDUA Gene Studied in Iranian Population
IDUA variants reported according to cDNA Refseq NM_000203.3 and protein Ref Seq NP_000194.2
Novel variants detected in the current study are marked in bold.
Del, deletion; dup, duplication; fs, frameshift.
Discussion
To the best of our knowledge, this has been the first comprehensive investigation of the IDUA mutation spectrum among Iranian families, containing data derived from the two previous studies conducted on Iranian patients with MPS1. In our population, 33 mutant alleles were found in 17 patients. The clinical features, enzyme activity, and genotypes of the patients are listed in Table 3. Only p.W402*, p.M1L, and p.L123R were found as being recurrent variants, with the p.W402* mutation being relatively common with a mean frequency of 17.6%, and p.M1L and p.L132R both emerged at the equal proportion of 11.7%. The p.W402* is the most commonly reported mutation underlying MPS1 worldwide. It is present in almost half of the mutant alleles in central and Western Europe and is also considered as the most prevalent mutation in North and South America and Australia (Bunge et al., 1994; Gort et al., 1998; Beesley et al., 2001; Bertola et al., 2011). In our neighboring country, Turkey, 43% of mutant IDUA alleles harbored this mutation, making it more frequent than that of our population. Although the frequency of p.W402* among Iranian patients was much lower than that of the Caucasian population, it is still the most common mutation.
Clinical Features and Genotypes of Iranian Mucopolysaccharidosis 1 Patients
All the information obtained in this study are shown in bold.
—, not mentioned; ND, not detected.
Another nonsense mutation, p.Y343*, which has been rarely reported so far (Lee-Chen and Wang, 1997; Voskoboeva et al., 1998), was found in a patient. As nonsense mutations result in a premature stop codon and consequently a truncated protein with no enzyme activity, it is plausible that four of screened homozygous patients carrying nonsense mutations (one with p.Y343* and three with p.W402*) presented the phenotype of Hurler syndrome as it is in agreement with previous studies in terms of their effects on phenotype (Terlato and Cox, 2003).
Two of the patients had a c.1A>G at the first base of the start codon in a homozygous state. This mutation impairs the translation of the start codon, yielding the loss of 133 amino acids from n-terminus of the alpha-l-iduronidase enzyme. The next initiation codon acts in place of the standard start codon for translation initiation, thus leading to a truncated protein in which three first exons have been eliminated. Moreover, this substitution gives rise to the deletion of n-terminus signal peptides essential for the sorting of protein to endoplasmic reticulum and lysosome and consequently disrupts the transport process involved in protein localization (Lee-Chen and Wang, 1997). Despite carrying the same start codon mutation, the patients, Nos VIII and V, indicated mild and intermediate phenotypes, respectively. This mutation has been previously observed in Turkey, China, and Spain (Lee-Chen and Wang, 1997; Bertola et al., 2011; Atceken et al., 2016). Other known missense changes, found in Iranian patients were p.G51D and p.P533R that have previously been reported in patients with different nationalities. The p.P533R is considered a common deleterious variation in North African countries) Laradi et al., 2005; Chkioua et al., 2007, 2011a, 2011b; Tebani et al., 2016), which is also present in the Mediterranean area, reaching up to 42% of mutant alleles in the island of Sicily (Gatti et al., 1997). Considering the distribution pattern of p.P533R, it is believed that this allele has probably spread from Africa to Mediterranean countries with the expansion of Islamic territory (Botigué et al., 2013).
An Italian study discovered p.G51D as a founder mutation with a low frequency of 11%, and later it was found to be present in Spanish (2.5%) and Italian populations (12%) in a comprehensive screening test aimed at 102 European patients (Bertola et al., 2011). This variant accounted for 8% of mutant alleles.
In addition to the two previously identified IDUA variations in which nucleotide changes resulted in amino acid substitutions (p.L132R and p.W175R) in Iranian patients (Yassaee et al., 2017), this study has led to the identification of additional ones (p.D301E, p.G134V, and p.S157P), all of which are highly likely to be pathogenic. The Aspartic acid 301 was predicted to be an active site residue. Substitution of highly conserved Asp301 by a glutamic acid in the active site of the enzyme would adversely affect enzyme activity (Bie et al., 2013). Substitution of serine by bulkier and more hydrophobic amino acid proline at position 157 would most likely disturb flexibility of the helix and perturb helical structure.
Multiple sequence alignment of IDUA across various species demonstrated that the three residues affected by missense changes are highly conserved, suggesting that these sites are critical to function and structure of the enzyme (Fig. 3). In silico analysis was carried out using SIFT, Polyphen2, mutation taster, and Mutpred to further assess the functional impacts of these amino acid variations on protein. SIFT predicted the three novel variations (p.D301E, p.G134V, and p.S157P) as being tolerated. Mutation taster called p.D301E and p.G134V as disease causing, but p.S157P was predicted to be polymorphism. Polyphen2, Mutpred, and PHD-SNP called the three missense changes probably damaging, highly harmful, and disease, respectively.

Multiple sequence alignment of the alpha-l-iduronidase protein from various species showed that the three mutated residues G134, S157 and D301 are highly conserved among different species. Amino acid sequences of different species were driven from NCBI Refseq database: Homo sapiens (NP_000194.2), Gallus gallus (NP_001026604.1), Castor Canadensis (JAV43677.1), Pan troglodytes (PNI30769), Canis lupus familiaris (NP_001300812.1), Rattus norvegicus (NP_001165555.1), Mus musculus (AAH57935.1), Sus scrofa (JAA74263.1), Macaca mulatta (AFE73120.1), Callithrix jacchus (JAB28983.1), Felis catus (NP_001291961.1) (Canis lupus familiaris) NP_001300812.1. The mutated amino acids are shown in black boxes.
It is noticeable that in a group of five patients from the southern part of Iran who were examined by Nasr-Esfahani et al. (2007), two patients presented with the same novel mutation (p.L132R). This variation was also observed earlier in this region but not anywhere else in Iran, suggesting that p.L132R is confined to this region and the patients share a common distant ancestor, although they belong to unrelated families.
Apart from single nucleotide changes identified in our population, a deletion and an insertion were also observed, including in patient XI and in patient XIII. In a previous study, a patient (Nos XI) revealed a four-nucleotide duplication c.612_615dupCTGC (p.S206Lfs*194) in exon 5 (Yassaee et al., 2017). This variant triggers stop codon 194 amino acids from codon 206, leading to deletion of 253 amino acids from the c-terminus of the protein. Interestingly, IDUA mutation c.704ins5 (c.613_617dupTGCTC), which is the second most commonly observed mutation in both Japanese and Korean populations (Poletto et al., 2018; Yamagishi et al., 1996; Kwak et al., 2016), is closely located to the newly detected duplication c.612_615dupCTGC in Iranian patients.
Patient XIII had a large deletion of 35 bp (c.590-17_607del35), starting at the end of intron 5 and extending 18 bp into exon 6 that resulted in the loss of splice acceptor site for exon 6. In silico analysis tools demonstrated that the deletion disrupts the original splicing sequence and most probably affects pre-mRNA splicing. The patient with this mutation showed a severe form of MPS1 and unfortunately deceased at 6 years of age as a consequence of pulmonary infection. To date, six deleterious disease-associated variants have been reported at this position, including p.G197S, p.G197D, p.Y201*, p.Y202*, p.D203N, and IVS5-7G>A. In addition, other variants (p.C205Y, p.G208D, and p.G208V) are closely located to the c.590-17_607del35.
In one patient, a mutant allele remained unidentified. First, it is because we did not screen all regions of the gene such as deep intronic regions and promoter sequences, where unidentified mutations could exist. Second, the Sanger sequencing method fails to identify large rearrangements and deletions.
Genotype-phenotype correlations were determined for individuals in whom both IDUA mutant alleles were detected. Nonsense mutations in homozygous state are known to cause classic Hurler phenotype (Scott et al., 1992; Bunge et al., 1994; Gort et al., 1998; Voskoboeva et al., 1998; Li et al., 2002), which was also the case in our patients with homozygous nonsense mutations, p.W402* and p.Y343*. In this study, a patient homozygous for p.M1L presented Hurler-Scheie, while the other patient with the same variation exhibited a mild form of the disease (Scheie syndrome). The reason for this phenotypic variation is that the severity of MPS1 disorder is not only determined by genotype but also by other risk contributors, such as environment and gene modifiers, which indirectly influence clinical features. A patient displaying the p.S157P homozygous genotype had rapidly progressive phenotype of the disease as well as severe cognitive impairment. As patient VI, who was homozygous for p.GFLNYY197-202, also showed a rapidly progressive phenotype, we suggest that this newly identified mutation must be severe too. The novel synonymous variation p.D301E that is located in the active site of the enzyme resulted in Scheie syndrome.
Up to now more than 200 different disease-causing lesions have been reported in the literature for the IDUA gene. With the exception of a few globally frequent mutations in the IDUA gene, many populations, even with the same ethnic background, show a different mutational pattern. Of the two common mutations reported until now in Caucasian (p.W402* and p.Q70*), p.W402* was more frequent than any other variations identified across all patients studied in Iran so far, but p.Q70* was absent from our population. p.Q70* appeared in 54% of mutant alleles (Bunge et al., 1994) in Russia and showed its highest frequency in Scandinavia, contributing to 54% and 78% of alleles in Norway and Finland, respectively (Voskoboeva et al., 1998; Chistiakov et al., 2014). The prevalence of p.Q70* decreases as one moves from northeast to southwest, becoming less prevalent in Poland (30%), Austria (31%), and Italy (14%). In fact, there is a geographic gradient in the distribution of Q70* from the north where it is commonly present, to the south where it rarely appeared. Additionally, the most frequent pathogenic mutations which occur in East Asian population, including p.Arg89Gln, p.Leu346Arg, c.1190-1G>A, and c.613_617dupTGCTC (Yamagishi et al., 1996; Lee et al., 2004; Sun et al., 2011; Wang et al., 2012; Kwak et al., 2016) were not detected within our population.
Although some of the common mutations found in our population, such as p.W402* and p.P533R, were encountered in patients from the Middle East (Chkioua et al., 2011a, 2011b; Cobos, 2015), none of the other pathogenic variations seen in our population was present in patients from other regions of the Middle East. Available data also show that there is a considerable allelic heterogeneity in the Middle Eastern population in which no founder mutation was present (Beesley et al., 2001). Therefore, it is suggested that comprehensive mutation screening of IDUA should be considered for people of Middle-Eastern origin suspected of having MPS1.
In conclusion, the present study illustrates the mutation spectrum of the IDUA gene in a cohort of Iranian patients. We identified 10 sequence variants, of which 4 were novel. Along with the other seven variations detected by previous studies, it was noted that despite the identification of a number of previously known variants, nearly half of the variations were unique in Iranian patients. Our results expanded the molecular and clinical information that will assist physicians in better treatment options as well as higher accuracy in the diagnosis of the disease, and appropriate genetic counseling for patients and their at-risk relatives.
Footnotes
Acknowledgments
The authors gratefully thank Dr. Masoud Houshmand who provided blood samples for molecular analysis. They would also like to express their gratitude to Mrs. Ahoora Arasteh Kni and Mrs. Masoomeh Dehghan Manshadi for their valuable contribution throughout the duration of this survey.
Authors' Contributions
M.K. designed the experiment, conducted genotyping of the patients and analyzed the data obtained from the experiment and wrote the article. M.A. collaborated in recruitment of patients and healthy controls, data collection, and interpretation. He also assisted in writing and editing the article.
Author Disclosure Statement
No competing financial interests exist.