
Abstract
Aims:
Cleft lip with or without cleft palate (CL/P) is a common birth defect with an average prevalence of 1/700 to 1/1000. Almost 70% of CL/P cases are nonsyndromic CL/P (NSCL/P). The aim of this study was to identify the underlying cause of a four-generation Chinese family with autosomal dominant NSCL/P.
Methods:
Genomic DNA was extracted from peripheral blood leukocytes, and whole-exome sequencing was carried out to identify the underlying genetic cause of the disorder. The mutation was confirmed by Sanger sequencing and polymerase chain reaction-restriction fragment length polymorphism methods. Western blotting and coimmunoprecipitation were used to analyze the protein expression level and adhesive dimerization of the CDH1 mutants. Slow aggregation assays were conducted to investigate the cell-cell adhesion ability.
Results:
A novel missense mutation (c.468G>C/p.Trp156Cys) of CDH1 was identified in the proband and the mutation was shown to cosegregate with the phenotype in the family. Furthermore, we found that the p.Trp156Cys mutation led to decreased E-cadherin dimerization and cell-cell adhesion ability.
Conclusions:
Our findings identified a novel CDH1 variant (c.468G>C/p.Trp156Cys) responsible for NSCL/P in a Chinese family, which expanded the mutational spectrum of the CDH1 gene and may contribute to understanding the molecular basis of NSCL/P.
Introduction
Cleft lip with or without cleft palate (CL/P) is one of the most common facial birth defects, with the prevalence of 1/700 to 1/1000. CL/P mainly consists of three major subtypes: cleft lip (CL), cleft palate (CP), and cleft lip with cleft palate (CLP). Almost 70% of CL/P patients belong to nonsyndromic CL/P (NSCL/P) based on the absence of additional structural or cognitive abnormalities. NSCL/P can result in complications affecting feeding, speech, and hearing (Dixon et al., 2011). The etiology of NSCL/P remains largely unknown, and it is believed that genetic factors, environmental factors, or their combination are responsible for NSCL/P (Leslie and Marazita, 2013). Family history of clefts would increase the incidence rate of NSCL/P in newborns (Sivertsen et al., 2008), suggesting genetic factors play a crucial role in the development of NSCL/P. Until now, genome-wide association studies (GWASs) and GWAS meta-analyses in different ethnicities have identified at least 40 genetic loci associated with NSCL/P, including 1q32 (IRF6), 5p12 (FGF10), 8q24.21 (gene desert), 10q25.3 (VAX), and so on (Yu et al., 2017; Butali et al., 2019). In addition to the common nucleotide variants identified in unbiased GWASs, rare variants identified by whole-exome sequencing from family-based studies, such as CDH1, FGF8, FGFR4, TRPS1, and FTCD, could also confer risk to NSCL/P (Bureau et al., 2014). Taken together, multiple genes involved in epidermis/epithelium development and morphogenesis, and molecular pathways that include those involved with extracellular signaling factors, transcription factors, and cell adhesion molecules have been implicated in NSCL/P (Deshpande and Goudy, 2019).
CDH1 is highly expressed at key stages of orofacial development, as in the nasal and palate process regions between the fourth and fifth weeks, and at the lateral and medial nasal process region at the sixth week of human embryogenesis (Song et al., 2017). Studies have shown that CDH1 is a causative gene of CL/P (Hozyasz et al., 2014; Brito et al., 2015; Ittiwut et al., 2016; Cox et al., 2018), gastric cancer (Becker et al., 1994; Melo et al., 2017), and combined CL/P and cancer (Frebourg et al., 2006; Kluijt et al., 2012; Benusiglio et al., 2013; Obermair et al., 2019). Recently, variants in CDH1 and CTNND1 have been identified as the cause of blepharocheilodontic syndrome, of which CL/P is a variable feature (Ghoumid et al., 2017; Kievit et al., 2018).
In this study, we recruited and characterized a four-generation family with autosomal dominant NSCL/P and identified a c.468G>C (p.Trp156Cys) variant in CDH1 via whole-exome sequencing. Moreover, we found that the CDH1 c.468G>C mutation led to decreased E-cadherin dimerization and cell-cell adhesion ability, which might play a causative role in the pathogenesis of NSCL/P.
Materials and Methods
Subjects and genomic DNA preparation
A four-generation Chinese family with autosomal dominant NSCL/P was identified at the hospital of Xinxiang County, Henan Province in China. Informed consent was obtained from the participants of the family, and study protocols were approved by the Ethics Committee of Huazhong University of Science and Technology. For genetic testing, genomic DNA was extracted from peripheral blood samples as previously reported (Liu et al., 2008).
Whole-exome sequencing
Two micrograms DNA sample from the proband (III: 6) was sent to an external core facility (WuXi Apptec Co., Ltd.) for exome sequencing. A total of 51,646,629 reads with an average size of 100 bp were generated in the coding regions, exon-intron boundaries, untranslated regions, and flanking regions (±1 kbp), using Illumina hiseq2500 sequencer and Agilent SureSelect Human All Exon V5 (50M). The mean depth of base mapped to target region is 82.69 × and the coverage of sequencing depth is 89.29% sequenced at 20 × and higher. Nonsynonymous, splicing, and heterozygous variants with a minor allele frequency of ≤0.05 were filtered in Exome Aggregation Consortium (ExAC), Ensembl database and Genome Aggregation Database (gnomAD) to exclude polymorphisms, and the detrimental variants were screened using Mutation Taster, PolyPhen-2 and SIFT. The consequences of splicing-alterations were estimated by using NetGene2 Server.
Mutation analysis
Mutation analysis was performed via Sanger sequencing to screen the pathogenic variant. The candidate genes of the proband (III: 6) and his sister (III: 8, normal) were polymerase chain reaction (PCR)-amplified and sequenced to determine if they both carried the same variant. The primers used for Sanger sequencing are as follows: PVR_F: aaccatgccatcctgtaccct; PVR_R: ggtcacattcttgccgtccac; CDH1_F: cctccgtttctggaatccaa; CDH1_R: gctgtagaaaaccttgcctt.
Restriction fragment length polymorphism (RFLP) analysis was used to detect whether the mutation cosegregated with the disease in this family, and whether the mutation was absent in 211 normal controls.
A PstI restriction site was created in PCR fragment of CDH1 c.468G>C mutation by using site-directed primers. The exon 4 of CDH1 gene was nested PCR-amplified from members of the family as well as 211 unrelated healthy Chinese individuals using the following PCR primers: one set was amplified as the template for the next round of PCR (CDH1_F1: tgtcttatcttgttcctcatctt and CDH1_R: gctgtagaaaaccttgcctt, with amplicon-346 bp), and the other set was amplified for enzyme digestion (CDH1_F: cctccgtttctggaatccaa and CDH1_R1: gcagctgatgggaggaatact, with amplicon-101 bp). The 101 bp PCR product was digested with PstI (NEB) at 37°C for 12 h and then separated on a 4.0% agarose gel. PstI cleaved the mutant PCR product into 80 and 21 bp (migrated out of the gel), whereas the wild-type product could not be cleaved and only the 101 bp band could be observed after electrophoresis.
Plasmid construction
The full-length E-cadherin (NM_004360) cDNA sequence was amplified via RT-PCR from a cDNA library generated using HEK293 cells. We then cloned the full-length E-cadherin cDNA sequence into the pcDNA3.1 (+) vector and tagged at the COOH terminal of E-cadherin with Flag tag (Ec1-F) or with Myc tag (Ec1-M). Plasmids carrying missense mutants (Trp156Cys or Trp156Ala) were generated based on the WT plasmid using site-directed mutagenesis (Vazyme) and verified by sequencing.
Cell culture and transfection
The cells were cultured in antibiotic-free Dulbecco's modified Eagle's medium (for HEK293T) or Dulbecco's modified Eagle's medium F12 (for Chinese hamster ovary [CHO] cells) containing 10% fetal bovine serum (Gibco, Grand Island, NY) and incubated at 37°C in 5% CO2 in a humidified incubator. Transient transfection of cells with plasmid DNA was conducted using Lipofectamine 2000 (Invitrogen, Carlsbad, CA).
Western blotting and coimmunoprecipitation
CHO cells were cultured in a 35 mm diameter tissue culture dish at 37°C and then transfected with wild-type or mutant CDH1 tagged with Flag tag. The transfected cells were cultured for 24 h and harvested in cell lysis buffer for western and immunoprecipitation (IP) (Beyotime Biotechnology, Shanghai, China). The protein samples were separated by 10% SDS-PAGE and transferred to nitrocellulose membrane. The membrane was treated with a mouse monoclonal antibody against Flag (MBL, Nagoya, Japan) or a mouse monoclonal antibody against β-actin (Beijing Ray Antibody Biotech, Beijing, China), followed by a second reaction with goat anti-mouse IgG coupled to horseradish peroxidase.
Co-IP experiments were conducted as reported (Klingelhofer et al., 2002), the transfected HEK293T cells with the same genotypic E-cadherin, which tagged with Flag or Myc epitope, respectively, were mixed with 1:1 ratio and cultured for another 14 h in a 10 cm-diameter dishes. The cells were washed with phosphate-buffered saline (PBS) buffer with 1 mM CaCl2 for 10 min and suspended in cell lysis buffer for western blot and IP. After sonication and centrifuge at 12,000 g for 10 min at 4°C, the supernatant was subjected to IP by subsequent incubation with antibody against Flag and Protein A Sepharose (EMD Millipore Corp., Darmstadt, Germany). The samples were analyzed by western blotting with antibodies against Flag and Myc (Sigma, MO).
Slow aggregation assays
The slow aggregation assays were performed as reported (Debruyne et al., 2014). CHO cells were transiently transfected with wild-type or mutant pcDNA3.1 (+)-CDH1 with Lipofectamine 2000 for 24 h and then trypsinized into single cells. Each well of the 96-well plate was coated with 50 μL of agar solution (100 mg Bacto-Agar in 15 mL of sterile PBS), and 2 × 104 cells were added to each well. The plate was incubated at 37°C in a humidified atmosphere with 5% CO2 for 24 h. Aggregation was evaluated under a Nikon inverted microscope (Tokyo, Japan). The size of aggregates was depicted as the area of their horizontal projections (Vilchis-Nestor et al., 2019), and the mean size of cellular aggregates of CHO cells transfected with WT or mutant E-cadherin was calculated using ImageJ software. Data were analyzed using Origin 8.0 software.
Results
Clinical features
The identified family was a four-generation Chinese family with 43 members, 10 (2 died, 1 aborted) of whom were patients with CL/P (Fig. 1A). According to our investigation, all the patients had no other clinical syndromes but CL/P features, so they were characterized as NSCL/P. The proband of the family (III: 6 in Fig. 1A) showed bilateral CL, and his son (IV: 4) and daughter (IV: 6, died) showed bilateral CL with CP. The younger sister of the proband (III: 10) showed unilateral CL (right side), and her son (IV: 9) also showed unilateral CL (right side). The patients in the pedigree showed different severity of CL/P, but none of them presented, at the time of evaluation, diffuse gastric cancer or other cancer types. In addition, the eyelids, nails, teeth, and hair did not present features of blepharocheilodontic syndrome.

Identification of a novel mutation c.468G>C (p.Trp156Cys) in the CDH1 gene underlying NSCL/P in a Chinese family.
Mutation analysis
To identify the pathogenic variant for NSCL/P in the pedigree, whole-exome sequencing of the proband (III: 6) was conducted. Variant annotation and filtration detected a rare variant PVR c.G499A (p.Val167Ile) and a novel variant CDH1 c.468G>C (p.Trp156Cys), which led to a substitution of tryptophan residue to cysteine residue at position 156 (p.Trp156Cys) of the protein (Fig. 1B), and no mutations affecting splicing were identified. Further RFLP analysis showed that the c.468G>C variant was present in all the patients (III: 6, III: 10, and IV: 9) but not in the unaffected members (III: 8, III: 16) of the family (Fig. 1C) or in 211 unrelated healthy Chinese controls (data not shown), indicating that the c.468G>C mutation in CDH1 cosegregated with the disease. However, the PVR variant c.G499A was excluded because the variant did not segregate with the disease in the family by Sanger sequencing analysis (data not shown). Moreover, the mutation c.468G>C was verified as novel by retrieval of the 1000 Genomes Project database, ExAC, gnomAD and the Human Gene Mutation Database (HGMD).
The protein sequence alignment showed that the Trp156 amino acid residue of E-cadherin is highly conserved from zebrafish to human (Fig. 1D). The p.Trp156Cys mutation was predicted to be deleterious by PolyPhen-2 (score at 1.00, Fig. 1E), and SIFT (score at 0.00) (data not shown). These suggested that the amino acid Trp156 of E-cadherin might be very important for its functions.
Effects of E-cadherin Trp156Cys mutation on its protein expression level and dimerization
To explore the possible mechanism of p.Trp156Cys mutation of E-cadherin causing NSCL/P, we compared the protein expression level and dimerization of p.Trp156Cys mutation with wild type by western blot and co-IP. The artificial mutation p.Trp156Ala reported by Klingelhofer et al. (2002) was used as a control, which impaired the adhesive dimerization of E-cadherin. The results showed that the p.Trp156Cys and p.Trp156Ala mutation had no significant influence on the total E-cadherin protein expression level compared with wild type (Fig. 2A, B). However, the p.Trp156Cys mutation showed decreased amount of adhesive dimers the same as p.Trp156Ala (Fig. 2C, D), which suggested that the p.Trp156Cys mutation could impair E-cadherin adhesive dimer formation.
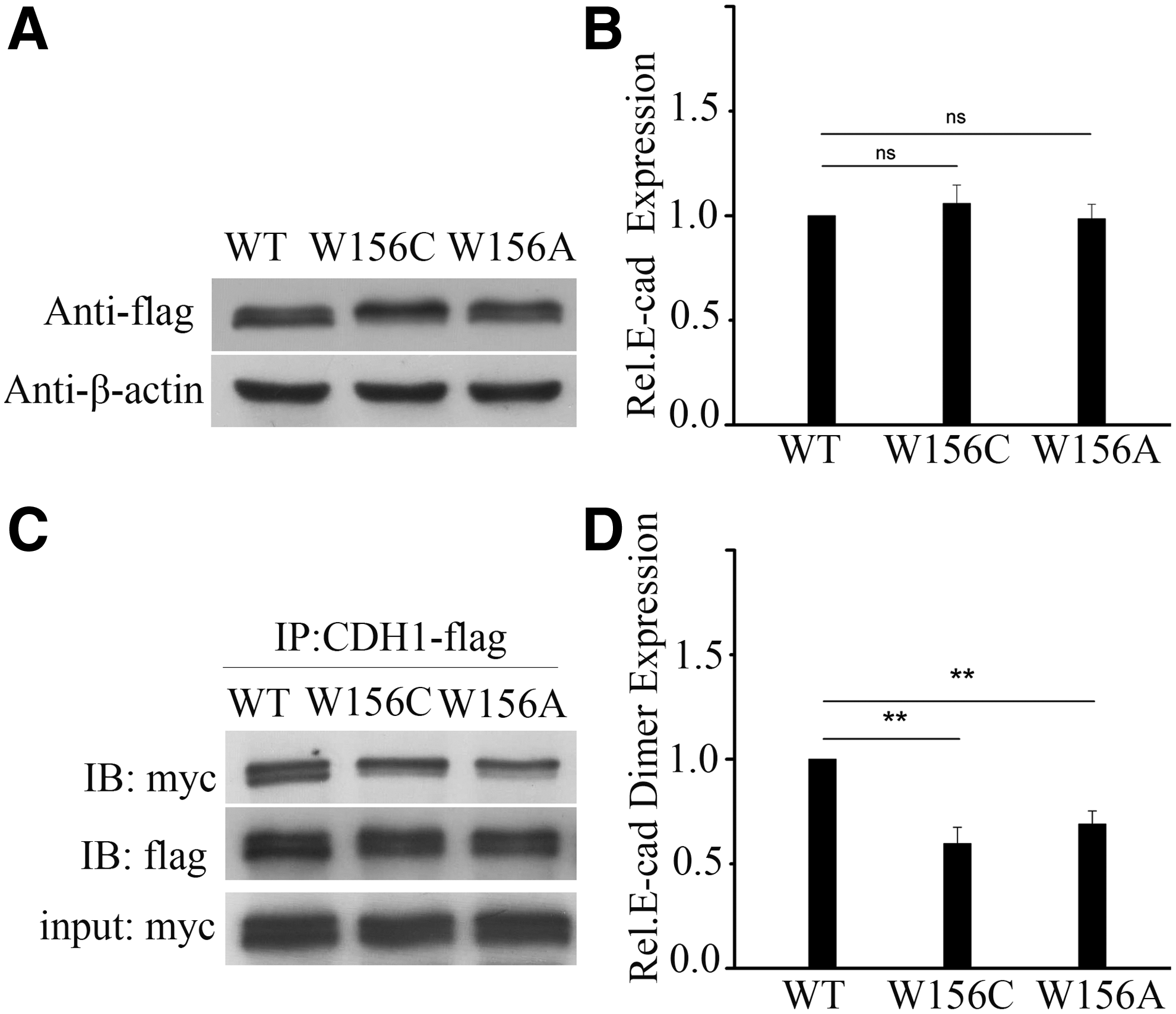
E-cadherin protein expression and adhesive dimerization for p.Trp156Cys mutation in CDH1.
Effect of E-cadherin Trp156Cys mutation on cell-cell adhesion ability
E-cadherin is the main component of the adherens junctions and is the major contributor to adhesion mechanisms, and E-cadherin dimers are essential intermediates in the assembly of adherens junctions (Shan et al., 2000). The above results showed that the p.Trp156Cys mutation in E-cadherin caused decreased E-cadherin dimerization, which suggested that it might impair cell-cell adhesion. To further explore whether the p.Trp156Cys mutation would affect cell-cell adhesion ability, cell-cell adhesion properties were assessed by slow-aggregation assays. We observed that wild-type CDH1-expressing cells showed large compact aggregates, while the cells expressing p.Trp156Cys or p.Trp156Ala mutation showed close-to-isolated phenotype, which was very similar to the phenotype of Mock cells (Fig. 3A, B). This suggested that the p.Trp156Cys mutation in E-cadherin could reduce cell-cell adhesion ability.
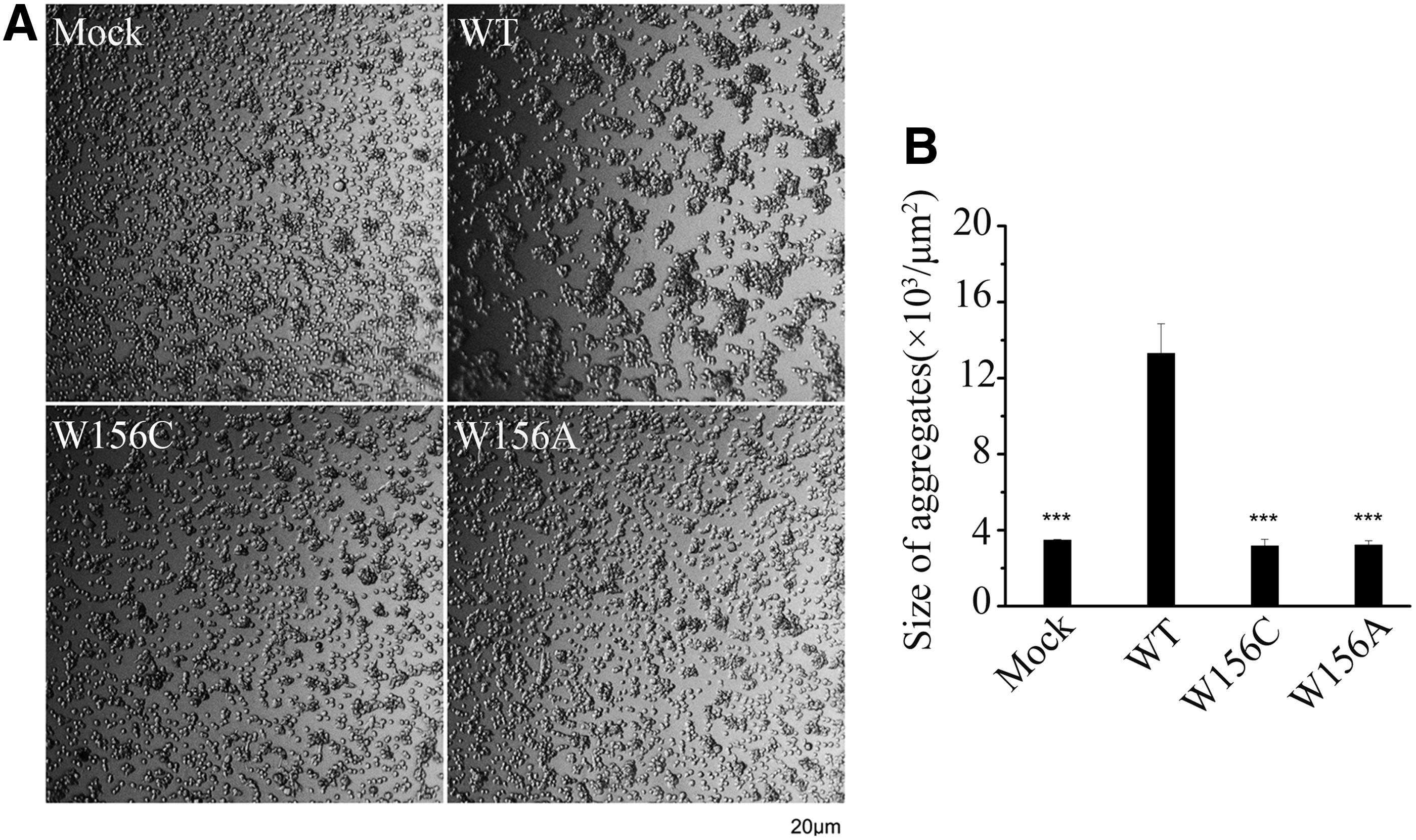
Discussion
The CDH1 gene encodes E-cadherin (OMIM: *192090), which is a transmembrane protein containing an extracellular domain, a single transmembrane domain and a highly conserved cytoplasmic domain. E-cadherin can form lateral (or cis-) and adhesive (or trans-) homodimers, and both types of dimers are essential intermediates in the assembly of adherens junctions (Chitaev and Troyanovsky, 1998; Shan et al., 2000). Adhesive dimers of E-cadherin are critical in calcium-dependent cell adhesion, and this type of dimerization is essential for the formation and maintenance of tissues and organs (Bruser and Bogdan, 2017). The E-cadherin dimerization was abolished when the tryptophan residue at position 156 of E-cadherin was changed to alanine (p.Trp156Ala) by biochemical characterization of cultured cells in vitro, which indicated that Trp156 of E-cadherin is important for its dimerization (Klingelhofer et al., 2002). Up to now, variants involving the amino acid 156 of CDH1 have not yet been associated with CL/P. The CDH1 nonsense mutations which generated premature termination codon at position 156 (p.Trp156*) were identified in somatic cells of stomach tumor (c.468G>A, data from Ensembl genome browser 96) and breast or ovarian cancer (c.467G>A, data from HGMD). Here, we revealed that a novel missense mutation c.468G>C (p.Trp156Cys) in CDH1 was associated with NSCL/P in a Chinese family, and none of the patients was found with cancers or family history of cancers until now (III: 6 aged 60 years; III: 10 aged 56 years and IV: 9 aged 26 years). Furthermore, we found that the p.Trp156Cys mutation did not decrease the expression of the protein, but impaired the dimerization of E-cadherin and cell-cell adhesion ability, which might contribute to the pathogenesis of NSCL/P in the Chinese family, while the reported nonsense mutations c.468G>A or c.467G>A (p.Trp156*) caused cancers probably because premature termination mutations might result in the degradation of E-cadherin.
As of now, 31 CDH1 mutations (including newly identified in this study) have been identified to be associated with CL/P or CL/P with cancer (Table 1) (Frebourg et al., 2006; Kluijt et al., 2012; Benusiglio et al., 2013; Vogelaar et al., 2013; Bureau et al., 2014; Brito et al., 2015; Ittiwut et al., 2016; Cox et al., 2018; Obermair et al., 2019). The variants in Table 1 are mainly distributed in the extracellular domain and clustered in the calcium-binding hinge regions between the extracellular cadherin (EC) domains. The p.Trp156Cys found in this study is located at the second amino acid of mature E-cadherin and at free N-terminus of extracellular domain (EC1), which mediates the binding of E-cadherin between cells. The variant types of CDH1 mutations in Table 1 contain missense, splicing, nonsense, and frameshift mutations. Among them, the CL/P with cancer was caused by splicing, nonsense, or frameshift mutations in CDH1 which might result in E-cadherin loss. Studies have shown that the loss of E-cadherin could result in impaired adheres junctions and anchorage-independence, which underpin the classical and typical invasion and metastasis formation in HDGC, and the inactivation of E-cadherin below a critical threshold would initiate the neoplastic process (Fitzgerald et al., 2010; Bruner and Derksen, 2018). Notably, all the missense mutations in Table 1 caused CL/P alone, among which some mutations, such as the mutation in a calcium-binding site p.Asp254Asn, were shown with decreased E-cadherin protein level, reduced location in plasma membrane or impaired adhesive ability (Brito et al., 2015). In this study, we found that the novel missense mutation p.Trp156Cys causing NSCL/P was due to decreased adhesive dimerization but not the total amount of E-cadherin. Thus, the findings in the study suggested that the variant CDH1 c.G468C may lead to NSCL/P by affecting the adhesive function of E-cadherin.
CDH1 Mutations Associated with CL/P or CL/P with Cancer
CL/P, cleft lip with or without cleft palate.
Interestingly, the second generation members in the pedigree showed total absence of CL/P phenotype, indicating the presence of incomplete penetrance. Also, the mutation carriers in the family showed phenotypic heterogeneity presenting bilateral CL (III: 6), bilateral CL with CP (IV: 4 and IV: 6), and unilateral CL (III: 10 and IV: 9) (Fig. 1A). The genetic association studies have found that some common and rare variants, such as SNPs in NOG, are significantly associated with cleft type differences (CL vs. CLP) and laterality differences (unilateral vs. bilateral) (Carlson et al., 2017). Notably, Epigenetics, such as CDH1 promoter methylation, may be a second hit to explain the patients with different cleft types (Alvizi et al., 2017). Moreover, the environmental factors the patients exposed to, such as maternal smoking, maternal alcohol consumption, and nutrients, have also been reported to increase the risk of the CL/P (Dixon et al., 2011). In the studies of other diseases, parental mosaicism and hypomorphic mutations are associated with the incomplete penetrance (Liu et al., 2008, 2019; Eshete et al., 2018). However, since DNA samples from the second generation members in the family could not be available, it is not possible in this study to examine the potential association between incomplete penetrance and CDH1 promoter methylation or other possibilities. By analyzing the exome sequencing data of III: 6, no other suspicious mutations were identified in the study. Furthermore, considering the incomplete penetrance was also observed in other NSCL/P families with CDH1 mutations (Brito et al., 2015), it suggests that the CDH1 variants alone do not seem to be the sole contributor to patients' phenotype in the family, and the effect of environmental factors and other unknown factors should be considered together to study the genotype-phenotype correlations in the NSCL/P disease.
In conclusion, we show that a novel CDH1 missense mutation (c.468G>C/p.Trp156Cys) is associated with NSCL/P in a Chinese family. Moreover, the mutation results in decreased E-cadherin dimerization and cell-cell adhesion ability, which may contribute to the pathogenesis of NSCL/P. These findings expand the mutation spectrum of CDH1 gene, which could offer a foundation to prevent beforehand of genetic disorders and contribute to a better understanding of the molecular mechanism of NSCL/P.
Footnotes
Acknowledgments
We thank the patients and their families for their participation in this study.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China grants (31671301, 31871262, 31701083, 31501019, 31400665); the National Key Research and Development Program of China (2016YFC1306000); and the National Science Foundation of Hubei Province of China (2017CFB692).