
Abstract
Background:
Hashimoto's thyroiditis (HT) is a common autoimmune disease characterized by lymphoid infiltration of the thyroid gland, including both T- and B-cells. Early studies have shown that HT is a complex disorder affected by both environmental and genetic factors. Recently, the single nucleotide polymorphism (SNP) rs2276886 associated with the CXCL9 gene was identified as associated with autoimmune thyroid disease susceptibility in Japanese populations. The aim of the present study was to validate this result for HT in a Chinese Han population.
Methods:
Study subjects, including 688 HT cases and 1456 healthy controls, were recruited, and 10 SNPs located within the CXCL9 gene were genotyped. Genetic association analyses were performed by fitting logistic models. Bioinformatics tools, including RegulomeDB and GTEx were utilized to investigate the functional consequences of the SNPs found to be significantly associated with HT.
Results:
SNP rs2276886 was identified as significantly associated with the risk of HT (odds ratio [OR] = 1.25, p = 0.0006). No significant expression quantitative trait loci (eQTL) signals could be identified for CXCL9. Significant eQTL signals were found for other genes, including ART3, CXCL10, CXCL11, NAAA, PPEF2, and SCARB2. This SNP physically maps to the CXCL9 gene region; however, further bioinformatic analyses indicated that this SNP might be associated with the gene NAAA.
Conclusions:
The rs2276886 SNP was found to be significantly associated with HT susceptibility. However, our findings suggest that this SNP which maps to the chromosomal region 4q21.1 likely effects the NAAA gene (as opposed to the CXCL9 gene), but still contributes to the susceptibility to HT in Han Chinese populations.
Introduction
As one of the autoimmune thyroid diseases, Hashimoto's thyroiditis (HT) is a common autoimmune disease that is characterized by lymphoid infiltration of the thyroid gland, including both T and B cells (Ajjan and Weetman, 2015). The etiology of HT involves excessively stimulated CD4+ T cells (Pyzik et al., 2015), and a decreased sensitivity of CD4+ T cells to the inhibitory effects of TGFβ may be another potential mechanism (Mirandola et al., 2011). Chronic inflammation in the thyroid gland during HT may impair thyroid function and cause hypothyroidism (depression), myxedema (heart failure), and goiter (painless swelling of the thyroid gland in the front of the neck). With this severe illness, few prevention measures can be prescribed for HT because of a lack of knowledge about its pathogenesis. Along with an elevation in the incidence of HT, studies on HT's pathogenesis have gradually increased.
Numerous studies have shown that the etiology of HT involves many factors such as genetic susceptibility (Akahane et al., 2016), diet (Xu et al., 2016) and infection (Caselli et al., 2012), but the true pathogenesis of HT has not yet been discovered. A higher sibling risk ratio in Korean (Villanueva et al., 2003) and German (Dittmar et al., 2011) populations suggests that there is a genetic predisposition to HT (Hwangbo and Park, 2018). With the rapid development of high-throughput sequencing, however, more and more susceptibility variants of complex diseases have been identified (Guan et al., 2014; Liu et al., 2014; Guan et al., 2016). However, current study results for HT account for only a small percentage of the estimated heritability in a few populations, and a corresponding biological interpretation is lacking and, therefore, the molecular mechanism of HT remains poorly understood.
Most recently, a single nucleotide polymorphism (SNP) (rs2276886) within the CXCL9 (MIG) gene was newly identified to be associated with HT susceptibility in Japanese populations (Akahane et al., 2016). Chemokines such as CXCL9 are a class of cytokine proteins that act as signaling molecules, regulating immune and inflammatory responses and modulating cell migration properties and localization of target cells such as leucocytes (Moser et al., 2004). The chemokine CXCL9, also known as MIG (a T cell chemoattractant monokine induced by IFN-g), which is commonly produced by local cells in inflammatory lesions, plays important roles in several autoimmune diseases (Smit et al., 2003; Tokunaga et al., 2018).
For example, CXCL9, which is highly expressed in the valvular tissue of chronic rheumatic heart disease patients, mediates the development of the disease by recruitment of T cells (Fae et al., 2013). Furthermore, increases in circulating CXCL9 have been found in patients with thyroiditis and hypothyroidism (Antonelli et al., 2011). Recently, Luo et al. (2018) found that increased levels of MRP14, which is induced by the IL-1beta/MAPK pathway, mediating TFC-derived chemokine secretion, including CXCL9 in HT patients, play a significant role in the mechanism of HT. Although many studies have hinted at the potential association between CXCL9 and HT, the underlying molecular mechanisms are largely unknown.
The polymorphisms of genes, including chemokines, have been conclusively identified as risk factors in the pathogenesis of HT (Akahane et al., 2016). A relationship between polymorphisms of CXCL9 and HT in the Japanese population has been shown (Akahane et al., 2016). However, the contributions of CXCL9 to the etiology and pathogenesis of HT have not yet been fully elucidated.
To date, the roles of CXCL9 in HT susceptibility in the Han Chinese population have not been investigated. Considering the genetic heterogeneity in different ethnic populations, the exploration of a potential association between the CXCL9 gene and HT among other independent populations is needed to confirm the previous results, which may shed light on its underlying mechanisms. Therefore, to determine whether the CXCL9 gene is associated with susceptibility to HT in Han Chinese populations, we conducted a hospital-based case/control study, which is the first study to evaluate the relationship between the CXCL9 gene and HT in a Han Chinese population.
Methods
Study subjects
Study subjects, including 688 HT cases and 1456 healthy controls, were recruited from Dongguan Tungwah Hospital between June 2011 and January 2019. Unrelated female individuals with a reported ancestry of Han Chinese were included in this study. Diagnosis of HT was based on (1) enlargement of the thyroid, (2) signs of hypoechogenicity and a nonhomogeneous texture on ultrasound images, and (3) a high level of either antithyroid peroxidase (TPO) or antithyroglobulin (Tg), with or without clinical and biochemical hypothyroidism. Individuals with a history of thyroid cancer and/or previous thyroid surgery were excluded from this study. Individuals with normal thyroid function and those who had negative results for thyroid autoantibodies were enrolled as controls.
Any individuals with the existence of any comorbid cardiac, autoimmune, infectious, musculoskeletal, or malignant disease, or a recent history of operation or trauma were excluded from this study. Thyroid function tests were performed using The Roche Diagnostics COBAS 6000 E601 Module Immunochemistry Analyzer (Roche, Basal, Switzerland), and five indicators, including namely, free triiodothyronine, free thyroxine, thyrotropin, anti-TPO, and anti-Tg, were measured in this study. Demographic information such as age, family history of HT, smoking, and alcohol drinking status were collected based on structured questionnaires. Peripheral blood was obtained from each study subject. This study protocol was approved by the Medical Ethics Committee of Dongguan Tungwah Hospital. Written informed consent was obtained from all of the participants.
SNP selection and genotyping
Based on 1000 genomes of Chinese Han Beijing data, SNPs with a minor allele frequency (MAF) ≥0.05 located within the gene region of CXCL9 were included in this study. A total of 10 SNPs were selected for genotyping. We extracted the genomic DNA from peripheral blood leukocytes using the Genomic DNA Kit (Axygen Scientific, Inc., CA). The Sequenom MassARRAY RS1000 system (Sequenom, San Diego, CA) was used for SNP genotyping. Typer Analyzer software was utilized for genotype data processing (Guan et al., 2012). Cases and controls were blind labeled during the genotyping process. Five percent of our samples were randomly chosen for replicating and the genotypes were reported to be 100% the same as in the previous sequencing run.
Statistical analyses
Logistic models were fitted for each SNP to examine their potential genetic association signals. Age was included as a covariate. In addition, we have also analyzed the potential association between the significant SNP and the thyroid function indicators. Linear models were fitted. Plink (Chang et al., 2015) was utilized for both logistic and linear regression analyses. Linkage disequilibrium (LD) structure was estimated using Haploview (Barrett et al., 2005). Haplotype-based association analyses were performed using Plink (Chang et al., 2015). Multiple comparisons were corrected by Bonferroni and the threshold of P values used for genetic association analyses was 0.05/10 = 0.005.
Bioinformatics analyses
Bioinformatics tools were utilized to investigate the functional consequences of the significant SNPs identified from the genetic association analyses. RegulomeDB (Xie et al., 2013) was used to examine the effects of the SNP on gene expression regulation. RegulomeDB is a publicly available database that annotates the genetic variants by integrating data obtained from the ENCODE project. It graded each SNP for its potential role played in the regulation of gene expression. In addition, expression quantitative trait loci (eQTL) data in multiple human tissues were extracted from the GTEx database (GTEx Consortium, 2013) for targeted SNPs to analyze their potential effects on gene expression.
Results
Demographic information
The demographic information about our study subjects is summarized in Table 1. No significant differences were obtained for age, smoking, or alcohol drinking status between the HT cases and healthy controls. Significant differences between cases and controls were identified for a family history of HT and all of the five thyroid functional indicators.
Characteristics of and Clinical Information About Our Study Subjects
FT3, free triiodothyronine; FT4, free thyroxine; HT, Hashimoto's thyroiditis; Tg, thyroglobulin; TPO, thyroid peroxidase; TSH, thyrotropin.
Genetic association
All of the selected SNPs had an MAF greater than 0.01 and were in Hardy-Weinberg equilibrium in our study subjects (Supplementary Table S1). We conducted Hardy-Weinberg equilibrium tests for each SNP in control samples. Among all of these 10 SNPs genotyped in our study, only SNP rs2276886 was identified to be significantly associated with the risk of HT (Table 2). As indicated, the A allele of SNP rs2276886 was found to be significantly associated with the increased risk of HT in our samples (odds ratio [OR] = 1.25, p = 0.0006), and a similar pattern with a significant signal was identified in the genotypic analyses (p = 0.0017). The full results of the association analyses are summarized in Supplementary Table S2.
Results of Genetic Association Analyses for Single Nucleotide Polymorphism rs2276886
Risk allele and significant p-values are in bold italics, and the threshold used for the significance of the p-value was 0.05/10 = 0.005.
OR refers to the risk allele odds ratio in both groups.
p-Values with adjustments for age.
CI, confidence interval; OR, odds ratio.
In addition, we have also performed association analyses for SNP rs2276886 and thyroid function indicators stratified by their HT status. No significant association could be identified between SNP rs2276886 and all five indicators in the HT cases and control groups (Supplementary Tables S3). The LD structure based on the genotype data is shown in Figure 1. One 2-SNP LD block (rs72607852-rs75180995) was constructed and results of haplotypic analyses were summarized in Supplementary Table S4. No significant haplotype was identified.

Linkage disequilibrium structure of the selected SNPs. Values of D’ are indicated in each square. The darker the color is, the greater the value. SNP, single nucleotide polymorphism.
Functional consequences and eQTL signals for SNP rs2276886
SNP rs2276886 is an intronic variant. We examined its potential functional consequences in RegulomeDB. RegulomeDB has a score system ranging from 1 to 7, and a lower score usually indicates more functional significance. SNP rs2276886 had a score of 1b, which indicated that it had significant functional significance. SNP rs2276886 was located at a DNaseI hypersensitivity region and was also found to be located at a transcription factor-binding region. We also examined the eQTL signals for SNP rs2276886 based on data extracted from the GTEx database. Although no significant eQTL signals could be identified for the gene CXCL9 (Supplementary Table S5), a couple of significant eQTL signals could be found for some other genes, including ART3, CXCL10, CXCL11, NAAA, PPEF2, and SCARB2 (Fig. 2). In addition, a significant eQTL signal for SNP rs2276886 in human thyroid tissue could only be found on gene NAAA (Fig. 3).
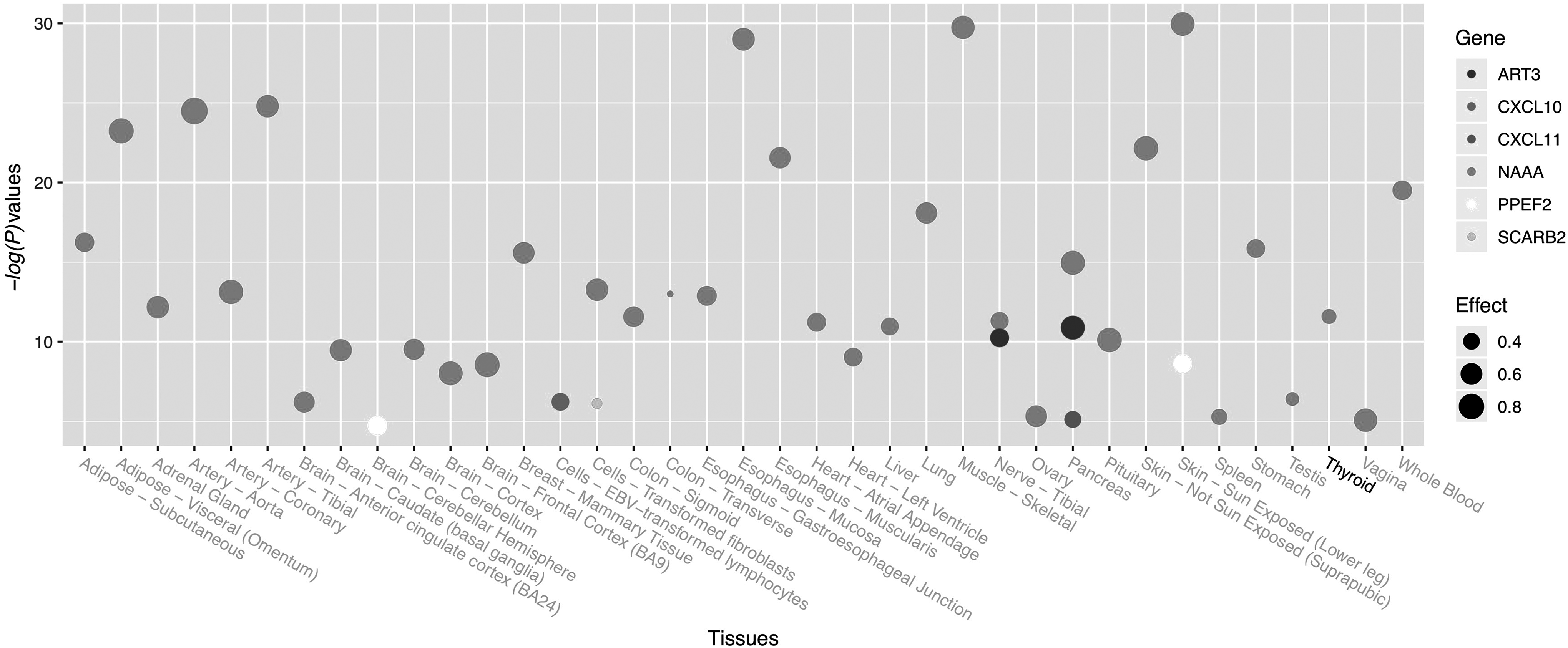
Significant eQTL signals for SNP rs2276886 on multiple genes. eQTL, expression quantitative trait loci.
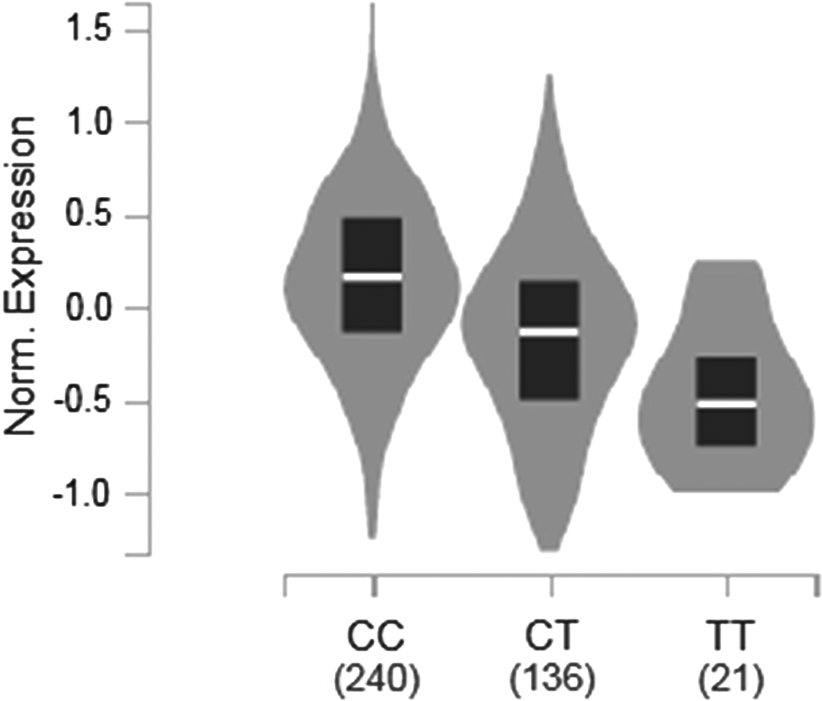
Expression levels of the gene NAAA in tissues of the thyroid for different genotypes of SNP rs2276886. Violin plots are based on the gene expression levels showed by the gray shade. The mean expression levels are showed by the white line, and 25% and 75% quantiles are showed by the black box.
Discussion
In the present study we have identified an intronic SNP rs2276886 to be significantly associated with HT. Our findings replicate those of a study conducted by Akahane et al., based on study subjects with Japanese ancestry. The A allele of rs2276886 was identified to be more frequent in patients in both studies. Considering the genetic similarity between Chinese Han and Japanese, this replication was not surprising. However, one interesting thing to note is our further eQTL analyses based on GTEx. Significant eQTL signals of rs2276886 were not identified for the gene CXCL9 but instead were found for some other genes in the 4q21.1 region, including ART3, CXCL10, CXCL11, NAAA, PPEF2, and SCARB2, which are located within a ±200 kb window of rs2276886.
We originally mapped SNP rs2276886 to the gene CXCL9 based on the physical location of this SNP (the SNP rs2276886 is located within the genetic region of CXCL9). This SNP gene mapping strategy based on physical location is a standard method used by most of the SNP genotyping microarray products and thus has been utilized by most of the previously published genome-wide association studies (GWAS). On the other hand, knowledge of eQTL signals might enable us to perform SNP gene mapping in a more accurate way. Instead of using the traditional mapping strategy based on physical location, we could map SNPs to genes based on their function (functional mapping), and thus the effect of SNP rs2276886 on the risk of HT was derived from a gene other than CXCL9.
Rs2276886 had a RegulomeDB score of 1b, which indicated that this SNP was located in a functionally significant genomic region and has a high possibility to be a SNP with functional consequences. Nevertheless, we need to be careful in interpreting these results based on bioinformatics analyses. As is known, a great many SNPs have significant eQTL data in GTEx simply because they represent points along an eQTL association peak.
We cannot simply consider the p-value as indicating that the SNP is a driver for the eQTL. Instead, we should further question whether the full range of eQTL data across a genomic range significantly colocalizes with a disease association peak. Functional studies in the future are needed to validate these eQTL signals. Thus, it is difficult to draw solid conclusions only from SNP-based association analysis (Yang et al., 2013; Chen et al., 2015; Guan et al., 2015; Zhang et al., 2015, 2018; Jia et al., 2016; Han et al., 2018), and further functional studies focusing on the associated SNP are desirable to elucidate the role of the SNP in the susceptibility of HT.
Among the six genes that showed eQTL signals for SNP rs2276886, NAAA is particularly interesting. Among 42 significant eQTL signals of rs2276886, 35 were obtained for NAAA from different human tissues, and what is more important is that a significant eQTL signal from human thyroid tissue was only identified for NAAA. In addition, SNP rs2276886 is located ∼50 kb upstream of NAAA, which is a hot spot for regulatory regions of genes.
NAAA encodes an N-acylethanolamine-hydrolyzing enzyme that hydrolyzes bioactive N-acylethanolamines to fatty acids and ethanolamine. The primary structure of NAAA exhibits 33-35% amino acid identity to acid ceramidase, which is a lysosomal enzyme hydrolyzing ceramide into fatty acids and sphingosine (Tsuboi et al., 2007). Previous GWAS have mapped NAAA to several human traits, including blood protein levels (Suhre et al., 2017; Sun et al., 2018), cerebrospinal fluid biomarker levels (Sasayama et al., 2017), and coronary artery calcified atherosclerotic plaques in type 2 diabetes (Divers et al., 2017). However, currently, there is no direct connection between NAAA and HT.
In the future, a thorough scan for genetic associations between polymorphic markers in NAAA and HT is necessary, and more biomedical research focusing on the pathological mechanisms of HT are needed.
Our study suffers from several limitations. First, only women were included in this study. Although this female-only strategy makes the sample enrollment process more efficient because of the high female:male ratio in HT cases, it in some ways hindered the generalization of the results obtained from this study. Studies with a more balanced gender distribution are needed in the future. Second, in the present study, only common polymorphisms were included. This strategy efficiently lowered the experimental cost but also dropped many low-frequency and rare variants.
In the future, a sequencing-based study is needed to thoroughly investigate the potential genetic architecture in the 4q21.1 region with regard to HT. Potential population stratification (PS) is another limitation of this study. As a common confounding factor in genetic association mapping, it might cause false-positive results. For the current candidate gene-based association study, we could not perform some correction methods such as principal component analysis or genomic control as used in the GWAS scenario. Considering that the Chinese population is a population with extensive genetic heterogeneity, the PS might be even more severe than we thought.
In summary, in the present study, SNP rs2276886 was identified to be significantly associated with HT susceptibility. The SNP was physically mapped to the gene region of CXCL9; however, further bioinformatics analyses have indicated that SNP rs2276886 might be connected to the regulation of the gene NAAA. Our findings suggest that 4q21.1 might be a susceptible locus for HT and rs2276886 may contribute to the susceptibility to HT in Han Chinese populations. Given the complexity of HT pathology and several limitations present in this study, functional studies based on experimental data and biomedical research focusing on the pathological mechanisms of HT are necessary in the future.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.