
Abstract
Background:
Familial exudative vitreoretinopathy (FEVR) is an inheritable retinal vascular disease, which often leads to severe vision loss and blindness in children. However, reported mutations can only account for 50-60% of patients with FEVR. The purpose of this study was to identify novel frizzled class receptor 4 (FZD4) and Norrin cystine knot growth factor NDP (NDP) mutations in a cohort of Indian patients with FEVR by whole-exome sequencing.
Methods:
We performed data filtering and bioinformatic analyses.
Results:
Two novel heterozygous mutations in FZD4 gene were identified, each in two different families: c.1499_1500del [p.500_500del] and c.G296C [p.C99S]. One novel mutation in NDP in another family was identified: c.A256G [p.K86E]. All FZD4 mutations affected conserved amino acid residues and were absent in 1000 control individuals. To assess the effect of these FZD4 mutations on the biological activity of the protein, we introduced each FZD4 mutation into FZD4 cDNA by the site-directed mutagenesis techniques. A Norrin/beta-catenin pathway-based luciferase reporter assay revealed that the c.1499_1500del failed to activate the luciferase reporter; in contrast, compared with the wild-type FZD4 protein, the, c.G296C [p.C99S] mutation exhibited increased luciferase reporter activity.
Conclusion:
Our study found two novel FZD4 mutations, with opposite effects regarding functional expression levels in Indian patients with FEVR and expands on the mutational spectrum of FZD4 in Indian FEVR patients.
Introduction
Familial exudative vitreoretinopathy (FEVR; OMIM: 133780) is an inherited, severe, retinal vascular disorder affecting the retinal blood vessel. FEVR patients present abnormal vascularization of the peripheral retina. Retinal folds and detachment often occur (Criswick and Schepens, 1969). The pathogenesis of FEVR starts from defective retinal angiogenesis in the peripheral retina. The resulting avascular peripheral retinal zone (Miyakubo et al., 1982; Shukla et al., 2003) is the hallmark of this disorder.
FEVR can be inherited in an autosomal dominant (adFEVR; OMIM: 133780), autosomal recessive (arFEVR; OMIM: 601813), or an X-linked recessive mode (OMIM: 305390) (Berger et al., 1992b). Genetic analyses have thus far identified 10 genes associated with FEVR: NDP (Berger et al., 1992a, 1992b), FZD4 (Meitinger et al., 1993; Robitaille et al., 2002; Toomes et al., 2004), LDL receptor-related protein 5 (LRP5) (Gong et al., 2001; Jiao et al., 2004; Salvo et al., 2015), tetraspanin 12 (TSPAN12) (Nikopoulos et al., 2010; Poulter et al., 2016), atonal BHLH transcription factor 7 (ATOH7) (Khan et al., 2012), zinc finger protein 408 (ZNF408) (Avila-Fernandez et al., 2015; Karjosukarso et al., 2018), kinesin family member 11 (KIF11) (Robitaille et al., 2014), RCC1 and BTB domain-containing protein 1 (RCBTB1) (Wu et al., 2016), catenin beta 1 (CTNNB1) (Panagiotou et al., 2017), and jagged canonical Notch ligand 1 (JAG1) (Zhang et al., 2020). Exudative vitreoretinopathy 3 (EVR3) has been mapped onto chromosome 11p12-13, but the causative gene has not yet been identified (Downey et al., 2001). Mutations in these genes can explain about 60% of FEVR patients. Among these genes, NDP, FZD4, LRP5, and TSPAN12 encode proteins in the evolutionary, conserved, Norrin/Wnt signaling pathway, which plays vital roles in retinal development and angiogenesis.
In this study, we applied whole-exome sequencing analysis to a cohort of FEVR patients in India and identified two novel FZD4 mutations and one NDP mutation. Functional studies revealed loss of the ability to induce luciferase reporter activity for FZD4 c.1499_1500del mutation and increased activity for FZD4 c.G296C mutation. Our data expand the mutation spectrum for FEVR patients in India.
Materials and Methods
Ethics statement
This study was approved by the Institutional Review Board of Sichuan Provincial People's Hospital and Aravind Medical Research Foundation. Written informed consent was obtained from each participant or their legal representative for minors. Thirty-two FEVR probands and their family members were recruited in this study based on the clinical diagnosis of FEVR. The diagnosis of FEVR was established based on ophthalmic examination and fundus fluorescein angiography, revealing at least one of the following classic findings: peripheral retinal avascularization, severe subretinal exudates, neovascularization, retinal fold or detachment, or vitreous hemorrhage. Patients with a gestational age of less than 38 weeks or a neonatal birth weight of less than 2000 g were excluded to eliminate interference of retinopathy of prematurity.
Whole-exome sequencing and variant confirmation
Genomic DNA was extracted from peripheral blood from study subjects using the DNA extract kit following the manufacturer's instructions (Tiangen, Beijing, China). Genomic DNA samples were fragmented, captured by the Agilent SureSelect Target Enrichment System (Agilent Technologies). Paired-end sequencing of enriched exome DNA fragments was performed using the Illumina HiSeq 2000 platform. After the sequencing platform generated the sequencing images, clean FASTQ data were obtained by CASAVA 1.82 (Illumina, Inc.). The clean reads were then mapped against UCSC hg19 by Burrows-Wheeler Aligner (BWA). Single nucleotide variants (SNVs) and insertions/or deletions (Indels) were detected by SAMTOOLS and noted by ANNOVAR (Zhou et al., 2015; Zhang et al., 2016). Low-quality variants and variants in noncoding regions without affecting splicing were excluded. Variants were filtered against the following databases: Database of Single Nucleotide Polymorphism (dbSNP), 1000 Genomes sequencing project, the gnomAD, and our 2001 in-house WES control samples' data. Nonsynonymous and splicing variants were filtered with MAF <0.01. The predicted functional effects of missense variants were ascertained through SIFT, PolyPhen-2, and PROVEAN predictors. Candidate mutations were confirmed by Sanger sequencing within families when DNA samples were available. Candidate variants were verified by direct Sanger sequencing analysis of genomic DNA samples from the probands and all available family members using primers flanking the variant sites (primer sequences are listed in Table 1).
Primers Used in This Study
Construction of expression plasmids
Expression plasmids carrying LRP5, FZD4, and Norrin cDNA were provided by Dr. Jeremy Nathans of Johns Hopkins University (Baltimore, MD) and previously described (Xu et al., 2004). FZD4 mutations identified in this study were introduced into the wild-type FZD4 cDNA using the QuikChange® Site-Directed Mutagenesis Kit (Agilent Technologies, Inc., Santa Clara, CA) following the manufacturer's instructions. Introduced mutations were verified by Sanger sequencing.
Luciferase reporter assays
Luciferase reporter assays were carried out as previously described (Xu et al., 2004). Briefly, 160,000 HEK293 STF cells/well were transfected with 800 ng DNA mix and 1.5 μL LipofectamineTM 2000 Transfection Reagent (Invitrogen, Carlsbad, CA) in 24-well plates. The DNA mix comprises 200 ng Norrin, 200 ng FZD4 (wild type or mutated), 200 ng LRP5, and 200 ng pSV-β-Galactosidase Control Vector. Forty-eight hours after transfection, transfected cells were washed with PBS three times and Luciferase reporter activity was assayed using the dual-Luciferase assay kit following the manufacturer's instructions (Promega, Madison, WI). Each assay was performed in triplicate. A representative result is shown from three independent experiments.
Expression study of FZD4 in cells
HEK293 STF and COS-7 cells (American Type Culture Collection [ATCC], Manassas, VA) were cultured in DMEM (Invitrogen, Waltham, MA) (with 10% fetal bovine serum and 1% (vol/vol) penicillin/streptomycin mixture). For western blotting assay, cells were cultured in six-well plates (Corning, Inc., Corning, NY) and transfected with 1 μg of wild-type or mutant pCMV6-FZD4 plasmid DNA using Lipofectamine™ 2000 at 50% confluency (Invitrogen). For immunofluorescence staining, COS-7 cells were seeded into a 12-well plate with a cover glass at the bottom of each well. Cells were transfected with 400 ng FZD4 plasmid (wild type or mutant) or 400 ng pCMV6 vector plasmid DNA. Forty-eight hours later, cells were harvested and used for immunofluorescence assay. After fixation with 4% paraformaldehyde, cells were stained with the anti-Flag antibody to monitor FZD4 protein expression.
Results
FZD4 mutation analysis
In this study, we used WES to identify causative mutations in a cohort of 32 FEVR patients in India. Two novel heterozygous mutations in the FZD4 gene were identified in four probands, accounting for 12.5% of all the families (Table 2). One is a frameshift deletion (c.1499_1500del [p.500_500del]) and the other is a likely deleterious point mutation (c.G296C, p.C99S) (Fig. 1). These two mutations were not reported in dbSNP, 1000 Genomes, gnomAD, or our in-house 2001 samples' WES databases. In addition, these two variants have not been previously reported in the literature and are absent from 1000 ethnicity-matched control samples.

Pedigrees of the four FEVR families with FZD4 mutations and DNA sequencing chromatograms.
Summary of Pathogenic Variants in FZD4 and NDP
NA, not available.
The first 2-bp deletion FZD4 mutation in exon 2, c.1499_1500del [p.500_500del], was found in two Indian families, FEVR-ICP-109 and FEVR-ICP-133. The proband of family FEVR-ICP-109 was a 7-year-old Indian male affected with the FEVR phenotype (Fig. 1a, c). His father was also affected. The proband of FEVR-ICP-133 was a 3.5-year-old Indian female affected with the FEVR phenotype and her father was also affected (Fig. 1b, c). This mutation causes a frameshift, resulting in incorrect amino acids after codon 499, followed by premature termination at codon 531. This variant affects a highly conserved region in the helix structure of the frizzled/smoothened family membrane domain (frizzled) of FZD4 and is predicted to be highly damaging (Fig. 1d-i). By protein secondary structure prediction and template-based tertiary structure modeling (Kallberg et al., 2012, 2014), we found that this mutation resulted in different conformation to the wild type (Fig. 1j, k).
The second heterozygous variant in FZD4, c.G296C [p.C99S], was identified in pedigrees FEVR-ICP-78 and FEVR-ICP-31. The proband of FEVR-ICP-78 was a 30-year-old female and her 28-year-old sister was also affected (Fig. 1e, g). The proband of FEVR-ICP-31 was a 14-year-old male and his 18-year-old brother and mother did not carry this mutant and showed the normal phenotype (Fig. 1f, g). This mutation changes the cysteine residue to serine at a highly conserved region in the frizzled domain (FRI) of FZD4 and is predicted to be damaging (Fig. 1h, i). By protein secondary structure prediction and template-based tertiary structure modeling (Kallberg et al., 2012, 2014), we found that this mutation also caused conformation change compared with the wild-type protein (Fig. 1j, l).
One novel mutation in NDP was identified in one family (Table 2): c.A256G [p.K86E] (Fig. 2a, b). This mutation also affected the highly conserved amino acid residue (Fig. 2c). K86 is localized in the conserved CT domain of NDP. This variant was not reported in dbSNP, 1000 Genomes, gnomAD, and our in-house 2001 samples' WES databases and is absent from 1000 ethnicity-matched control samples. This mutation likely affects protein conformation, as revealed by protein secondary structure prediction and template-based tertiary structure modeling (Kallberg et al., 2012, 2014).
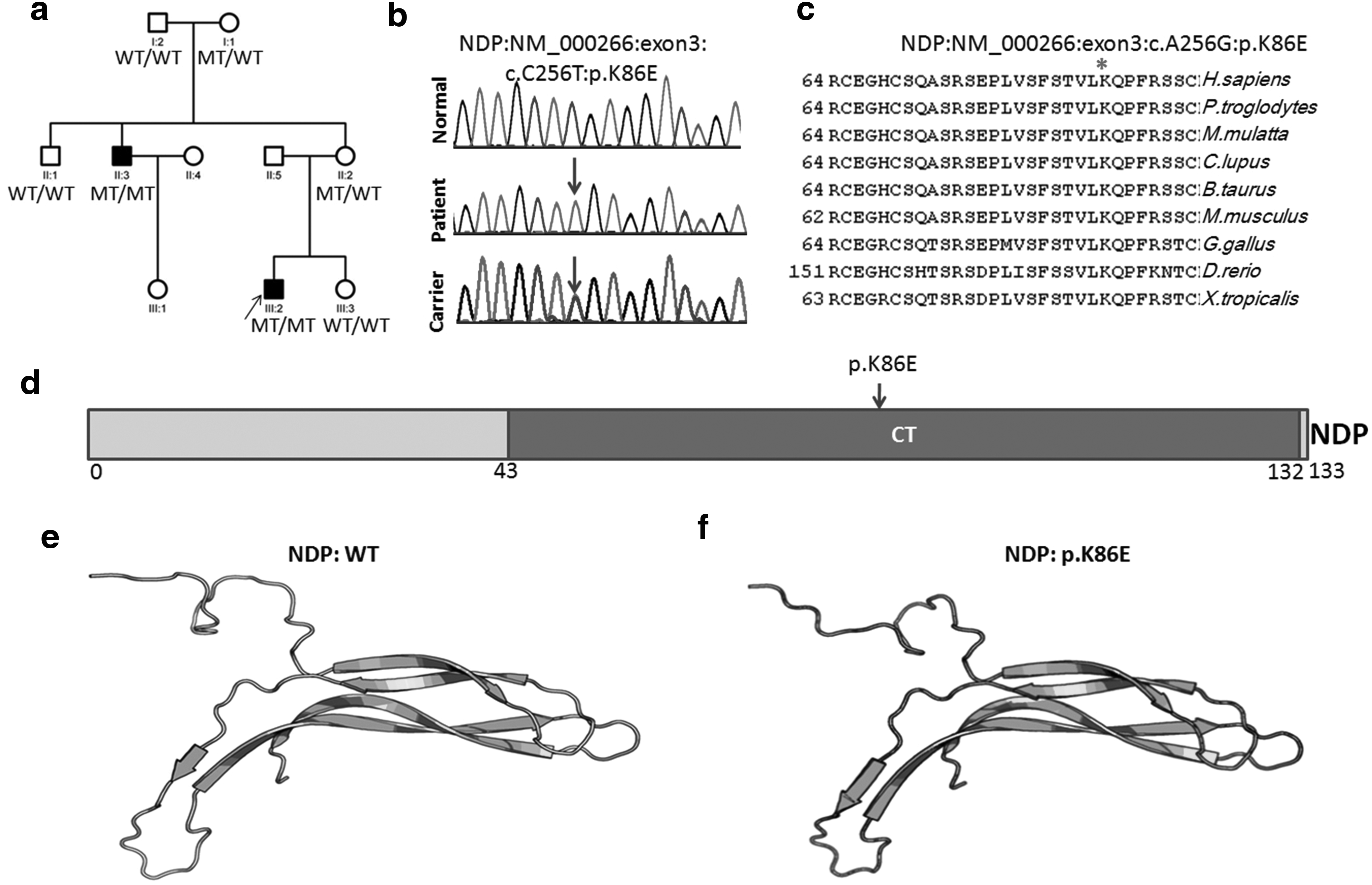
Pedigrees of the FEVR family with NPD mutation and DNA sequencing chromatograms.
Defective Norrin signaling mediated by mutant FZD4 proteins
To assess the impact of these two novel mutations on FZD4 biological function, we analyzed the function of mutant FZD4 proteins using a Wnt-responsive firefly luciferase reporter system. The FZD4 p.C99S mutant was expressed at the comparable level as a wild-type FZD4 protein (Fig. 3a). Under physiological conditions, a complex of Norrin, FZD4, and LRP5 activates canonical Norrin/β-catenin signaling. As shown in Figure 3b, both FZD4 mutation constructs produced significantly different TOPflash activity levels when compared with the WT FZD4 construct. The p.500_500del FZD4 mutant failed to mediate activation of the luciferase reporter in STF cells upon addition of Norrin (Fig. 3b). Interestingly, the FZD4 c.G296C [p.C99S] mutation displayed increased activity compared with the wild-type control. This result is similar to the CTNNB1 p.R710C mutation reported by Panagiotou et al. (2017). In their study, the CTNNB1 p.R710C mutation induced significantly higher levels of Wnt-responsive luciferase reporter activation. In addition, the NDP c.A256G, p.K86E mutation showed decreased luciferase reporter activation compared with the wild-type control (Supplementary Fig. S1).

Expression of FZD4 mutants and luciferase assays with the STF cell line transfected with plasmids expressing Norrin/catenin components.
Discussion
We applied WES analysis to identify causative mutations in a cohort of 32 unrelated FEVR families. Two novel mutations in the FZD4 gene were identified in four families (accounting for 12.5% of all the 32 families) and one mutation in one family in the NDP gene (accounting for 3.1% of all the 32 families). Previous reports have shown diverse mutation frequencies in these two genes; one main reason for this difference is due to a smaller sample size (Salvo et al., 2015). In a report of the Netherlands, eight (40%) and two (10%) mutations were found in FZD4 and NDP, respectively, in a total of 20 FVER families (Boonstra et al., 2009). In a target capture, followed by NGS analysis of a Chinese cohort, mutations in FZD4 were found in 15% of the probands (14/92) and 6.5% (6/92) probands were discovered with NDP mutations (Salvo et al., 2015). In a Canadian cohort report, the authors identified 11 mutations in 12 probands, with FZD4 mutations in 68 probands (17.7%) (Robitaille et al., 2011). The NDP gene caused 6% of FEVR cases in a Japanese population (Kondo et al., 2007). In an Indian cohort, mutations in FZD4 were observed in 5.6% of FEVR (Nallathambi et al., 2006).
As a member of the conserved frizzled gene family, FZD4 encodes a G protein-coupled receptor with seven-pass transmembrane domains and a frizzled (FZ) domain in the extracellular region for ligand binding (Fuhrmann, 2008). FZD4 is essential in Norrin/beta-Catenin signaling (Robitaille et al., 2002). In the Wnt-responsive TOPflash luciferase report assay, both p.500_500del and p.C99S FZD4 mutants induced significantly different reporter activity levels when compared with the WT FZD4 construct. The p.500_500del FZD4 mutant failed to mediate induction of luciferase reporter activity (Fig. 3b). On the other hand, the p.C99S mutation exhibited increased TOPflash activity. Similar TOPflash reporter activity results have been previously reported for NDP (Xu et al., 2004; Qin et al., 2008) and CTNNB1 mutations (Panagiotou et al., 2017). In the STF signaling assay, the NDP p.K58N mutant exhibited two-fold greater Norrin activity than the wild type (Xu et al., 2004). In addition, Ye et al. (2009) reported defective embryonic angiogenesis in mice with overactivated Norrin signaling. Recently, Panagiotou et al. (2017) reported that the CTNNB1 p.R710C mutation identified in a FEVR patient produced significantly higher levels of TOPflash activation.
Taken together, our study identified two novel FZD4 mutations and one NDP mutation in the Indian family with FEVR. These data provide valuable information to the mutation spectrum of FEVR.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was funded by the National Natural Science Foundation of China (81970841 and 81770950 to X.J.Z.) and the Sichuan Provincial Department of Science and Technology (2017TJPT0010, 2018YSZH0020, 2018JZ0019, and 2015SZ0060). The funding agencies had no role in study design, data analysis, or preparation of the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.