
Abstract
Background:
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related deaths worldwide. Patients suffering from HCC are usually diagnosed during an advanced stage, which limits the effectiveness of treatment. This phenomenon has led to an urgent need to discover promising HCC diagnostic biomarkers and to identify novel targets for HCC treatment.
Materials and Methods:
In this study, the gene expression profiles of the GSE45436 participants were downloaded from the Gene Expression Omnibus database. The HCC differentially expressed genes (HCC_DEGs) were identified through a comparison with healthy controls. The Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analyses were performed by DAVID, a free website used for annotating genes. Next, we used STRING, an online website, to identify likely protein-protein interactions among the DEGs. Cytoscape software was utilized to construct a protein-protein interaction network. MCODE, a plug-in of the Cytoscape software, was used for a module analysis. Finally, we used the Gene Expression Profiling Interactive Analysis website to determine the module genes' effects on overall survival.
Results:
A total of 313 genes were identified as differentially expressed, which comprised 118 upregulated genes and 195 downregulated genes. We used these data to identify 67 module genes. These were further verified using The Cancer Genome Atlas database resulting in 57 that remained statistically significant. Foremost, we identified one significant gene, DEP domain-containing protein 1B (DEPDC1B), which should be investigated for its usefulness as a new biomarker for diagnoses and prognoses.
Conclusion:
To our knowledge, DEPDC1B has not previously been reported as being associated with HCC. These results suggest that in silico methods, such as those employed, can provide valuable and even unique candidate biomarkers for further evaluation.
Introduction
Liver cancer is the second leading cause of cancer-related deaths worldwide, with ∼850,000 new cases each year. Hepatocellular carcinoma (HCC) accounts for ∼90% of all cases of primary liver cancer (Llovet et al., 2016). Tumor recurrence and metastasis are the primary reasons for the low survival rate of patients suffering from advanced liver cancer (Bruix, 1997). Liver cancer imposes a massive economic burden on health care. Chronic liver disease caused by hepatitis B and hepatitis C viruses together with alcohol overconsumption accounts for the majority of HCC cases (Stepanova et al., 2017). The incidence of nonalcoholic fatty liver disease has also been increasing and is also associated with the development of liver cancer (Starley et al., 2010). Although most cases develop in the context of background cirrhosis, the molecular pathogenic pathways vary among based on the etiological triggers (Ghouri et al., 2017). There is, therefore, a pressing need for the discovery of novel biomarkers to aid in earlier diagnoses of HCC. Currently, by analyzing serum alpha-alpha-fetoprotein levels in combination with imaging techniques, HCC can be diagnosed without histopathology. For the past decade, advances in genomic and proteomic platforms and biomarker assays have led to the identification of myriad candidate novel biomarkers that have improved the diagnosis of liver cancer. In this study, we analyzed the free National Center for Biotechnology Information-Gene Expression Omnibus (NCBI-GEO) database with bioinformatic tools, hoping to find valuable new clues.
The gene chip is a genetic-level detection technology that has been used in scientific research since 2000 (Kirby et al., 2007). The use of gene chip technology and bioinformatic methods can characterize the expression of the entire genome under a given set of conditions in a single experiment; thereby enabling the screening of differentially expressed genes (DEGs) based on clinical prevalence. Through the widespread use of this technology, a considerable amount of HCC expression data has been generated, archived, and stored in public databases (Kirby et al., 2007).
Materials and Methods
Data resource
Data from the Affymetrix microarray file GSE45436 was downloaded from the public NCBI-GEO database and executed on the GPL570 platform. GSE45436 comprises 93 tumor samples and 41 healthy controls. All samples in GSE45436 are processed by the robust multiarray average algorithm. All the specimens were collected before any chemotherapy (Wang et al., 2013).
Data preprocessing of HCC
After GSE45436 was downloaded, we transformed probe identification numbers into gene symbols. When multiple probes corresponded to one gene, the gene with the highest expression level is considered to be the final selected gene. Next, we used the limma package to identify HCC DEGs. p-Value <0.01 and |log2fold change (FC)| ≥ 2 as threshold.
Identification of HCC_DEGs
The raw data GSE45436 files used for analysis included the .CEL format files (Affymetrix). The limma package in R studio was applied to identify the DEGs between HCC and healthy control samples (Smyth, 2004). We used the linear model and eBayes functions of the limma package to test for differential gene expression. Genes with a |log2-FC| ≥ 2 and corrected p-values <0.01 were used as the cutoff criterion.
Gene Ontology and pathway enrichment analysis
To explore the biological functions of the aforementioned HCC_DEG, we utilized the DAVID online software to perform Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis (Ogata et al., 1999). DAVID is an analysis website for DEGs Annotation, Visualization and Integrated Discovery (Sherman and Lempicki, 2009). A false discovery rate of p < 0.05 was considered statistically significant.
Establishment of protein-protein interactions network
The STRING website was used to identify likely protein-protein interactions (PPI) (Franceschini et al., 2012). This database contains >24.6 million proteins and 2 billion interactions involved in 5090 organisms. We uploaded the HCC_DEGs onto the STRING database. The minimum required interaction score was set as the highest confidence >0.9. We used Cytoscape (version 3.6.1) software to visualize and analyze protein-protien interaction networks (Shannon et al., 2003).
Identification of hub genes
To identify important hub genes in these HCC_DEGs, the Cytoscape software plug-in of MCODE was used to filter important modules of the entire network (K = 12, other parameters are set to default values). Functional enrichment analysis of HCC_DEGs, with p < 0.05 was set as the threshold, to screen for the most important gene module.
Validation and survival analysis based on the The Cancer Genome Atlas database
The Gene Expression Profiling Interactive Analysis (GEPIA) online database was employed in the overall survival analysis (Tang et al., 2017). A logrank p < 0.05 was considered statistically significant. First, we used this online database for hub genes expression-level analysis, the GSE45436 and The Cancer Genome Atlas (TCGA)/Genotype-Tissue Expression (GTEx) liver hepatocellular carcinoma (LIHC) data sets. We set the parameters, |log2FC| cutoff: 1 and p-value cutoff: 0.01, as the threshold criteria, match TCGA normal and GTEx data set. Then, we conducted an overall survival analysis of the hub genes to observe the effect of the hub gene on the overall survival rate of LIHC patients. Qualified genes can be used as biological targets for diagnosis and treatment.
Result
Identification of HCC_DEGs
A total of 134 liver samples, including 93 tumor samples and 41 healthy tissues, were identified. After gene expression data processing and normalizing, we used the limma package to identify DEGs. A total of 313 DEGs were identified, including 118 upregulated and 195 downregulated genes from the GSE45436 data set (Table 1). A volcano plot and heatmap of the HCC_DEGs are shown (Fig. 1A, B).
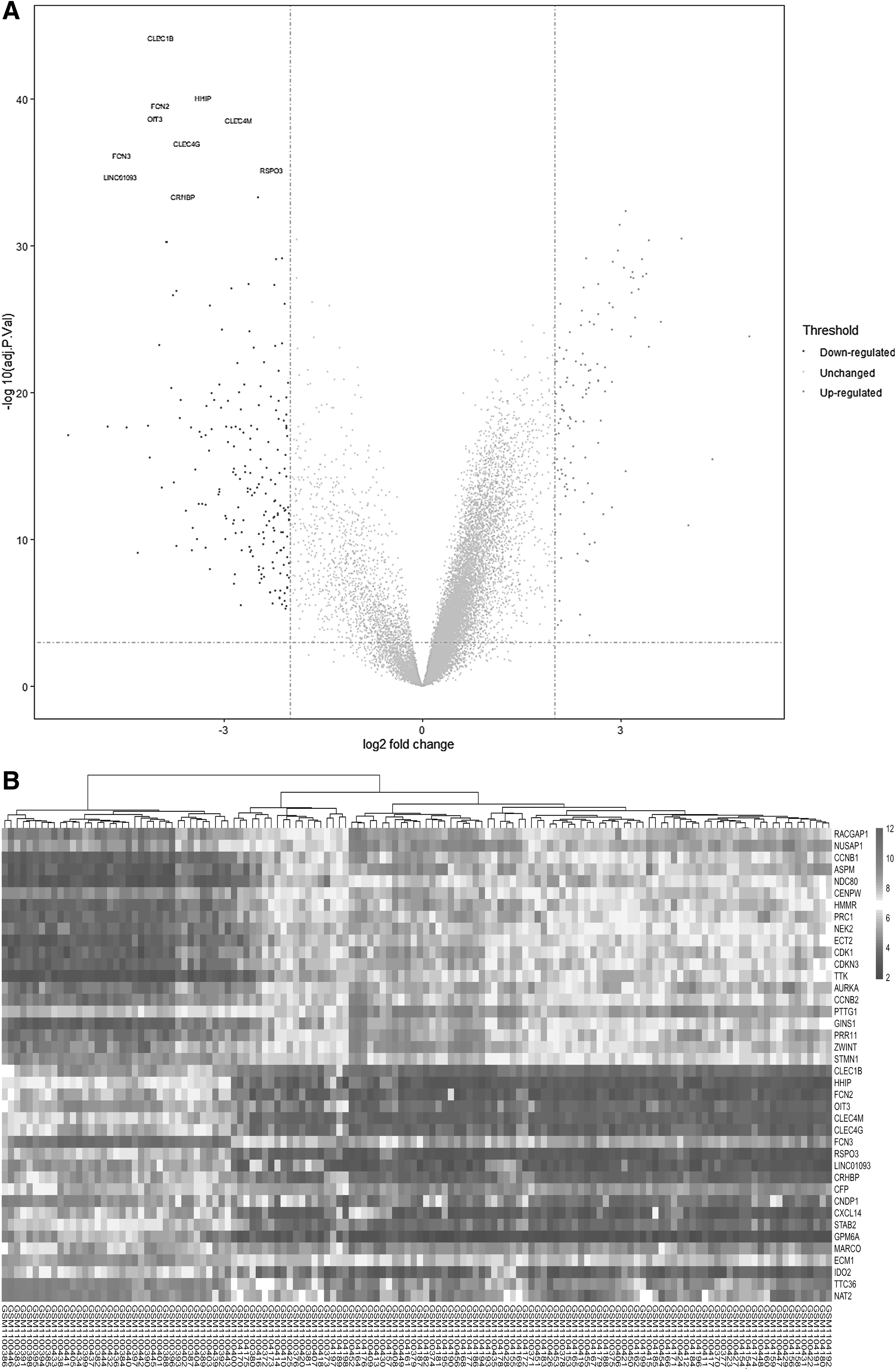
Volcano plot and heatmap of the DEGs (HCC_DEGs) between tumor tissues and carcinoma tissues from patients with hepatocellular cancer.
Three Hundred Thirteen Differentially Expressed Genes Were Identified in GSE43436, Including 118 Upregulated Genes and 195 Downregulated Genes
DEG, differentially expressed gene.
Gene term enrichment analysis and KEGG pathways analysis of HCC_DEGs
To investigate the biological functions of the aforementioned HCC_DEGs, an enrichment analysis of up- and downregulated DEGs was carried out. The HCC_DEGs functional enrichment analysis results were divided into three functional categories, including biological processes (BPs), cellular component (CC), and molecular functional (MF) (Table 2). For the BPs category, upregulated genes were enriched for mitotic cytokinesis, DNA replication initiation, regulation of cell cycle, positive regulation of cytokinesis, and cytokinesis. The downregulated genes were enriched for the bile acid biosynthetic process, the epoxygenase P450 pathway, complement activation and the complement classical pathway, cell chemotaxis, and
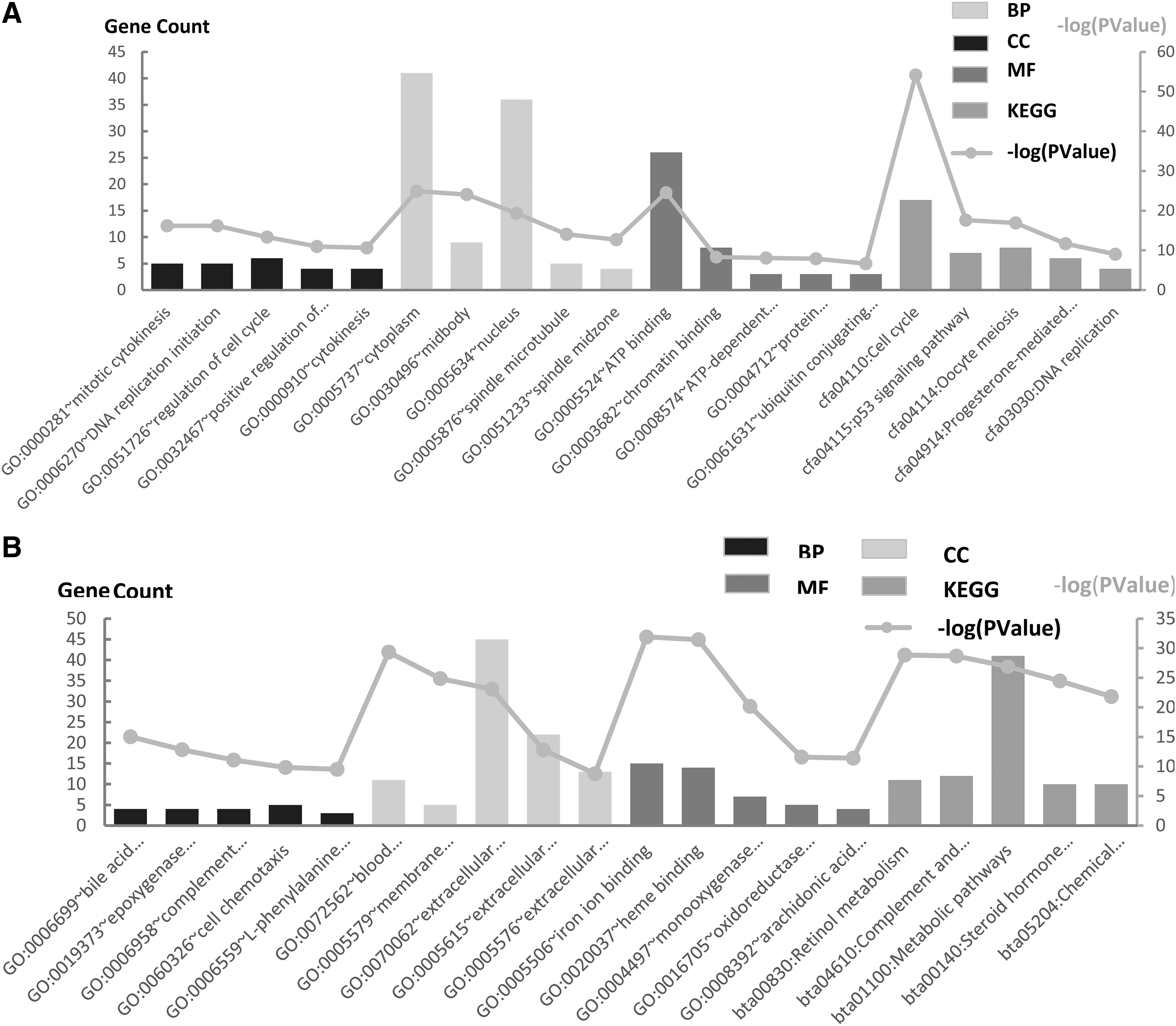
GO and KEGG pathway analysis of HCC_DEGs.
The Top Five Gene Ontology and Kyoto Encyclopedia of Genes and Genomes Enrichment of Differentially Expressed Genes
GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Establishment of PPI networks
All of the HCC_DEGs were uploaded to the STRING website to characterize hypothetical PPI networks. To visualize these PPI networks, we used Cytoscape. We identified a total of 272 nodes with 3125 edges through these analyses (Fig. 3).
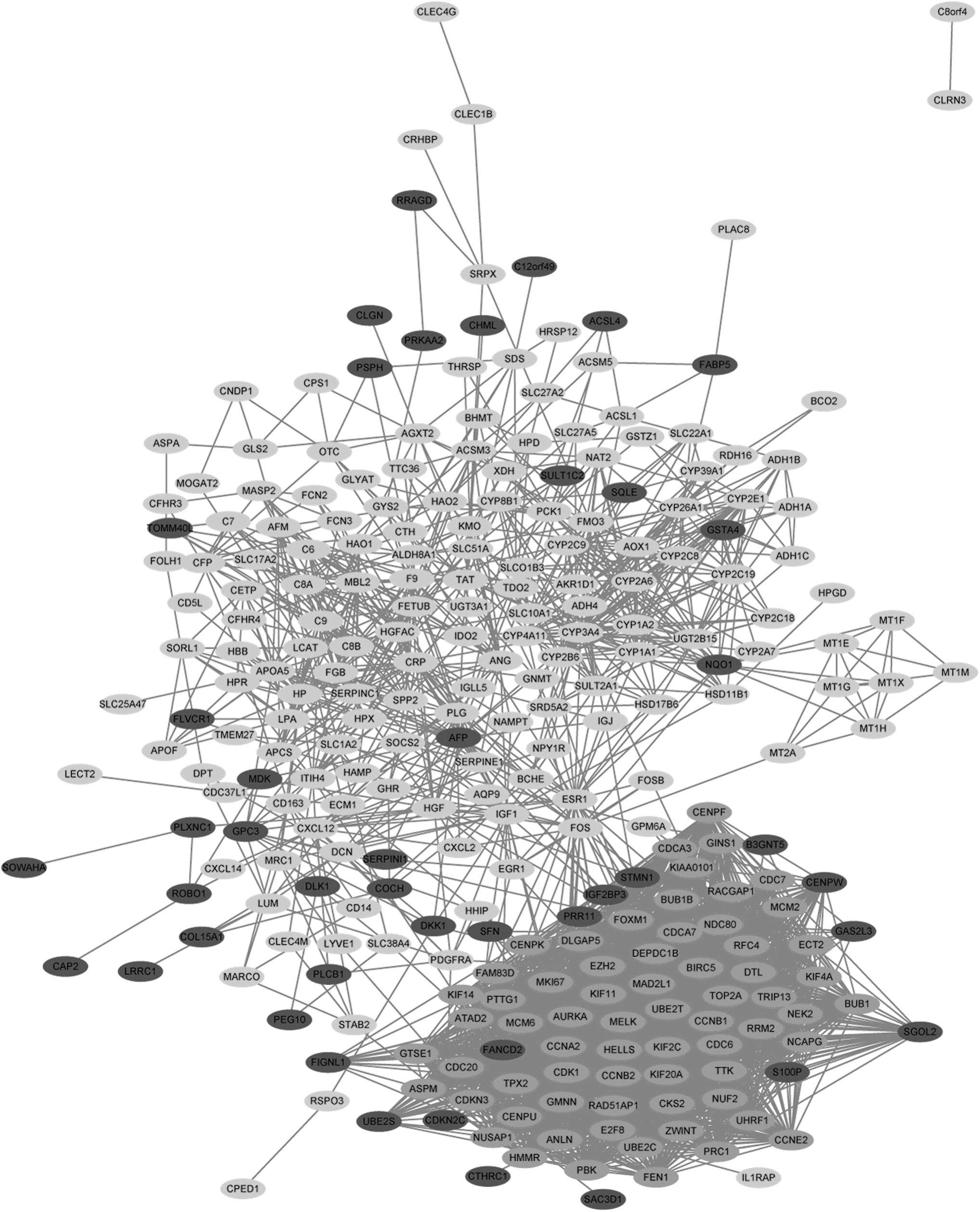
PPI network of HCC_DEGs. Dark gray means upregulated HCC_DEGs; light gray means downregulated HCC_DEGs. Medium gray means module genes. PPI, protein-protein interaction.
Module analysis and selection of hub genes from the MCODE network
One significant module was selected from the PPI network using the MCODE plug-in. This module comprises 67 nodes and 2008 edges (Fig. 4). HCC_DEGs in this module were significantly enriched for DNA replication initiation, midbody, ATP binding, and cell cycle (Table 3). These results indicate that the module genes are significantly associated with the pathogenesis of HCC. We used the MCODE function of the Cytoscape software to identify the hub genes from the entire PPI network. When K = 12 (K value represents the degree of clustering), other parameters are set to default values. We uploaded the module genes onto the TCGA database and found that 57 genes are associated with the prognosis of patients (Table 4). We tested the identified genes as molecular markers for diagnosis, treatment, and prognostic value.

The foremost module from the PPI network. Light gray means upregulated HCC_DEGs.
The Functional and Pathway Enrichment of Module Genes
The 57 Genes Are Associated with the Prognosis of Patients
GTEx, Genotype-Tissue Expression; LIHC, liver hepatocellular carcinoma; TCGA, The Cancer Genome Atlas.
Validation and survival analysis based on TCGA database
To assess whether gene expression changes were consistent between samples from the test GSE45436 and the validated LIHC (TCGA/GTEx) data set, we validated the hub genes (57 genes in total) on the GEPIA website. We found that in the LIHC data set (built into TGCA/GTEx), all genes were upregulated in tumors relative to normal liver cancer expression, which is consistent with the GSE45436 liver carcinoma sample (Fig. 5). For further verification, the total survival of each of the aforementioned 57 candidate genes was calculated, and the 57 genes were related to patients' prognostic (Fig. 6).

Gene expression level of prognostic-related biomarkers. Light gray represents the expression level of prognosis-related genes in tumor tissues, and dark gray represents the expression level of prognosis-related genes in normal tissues.
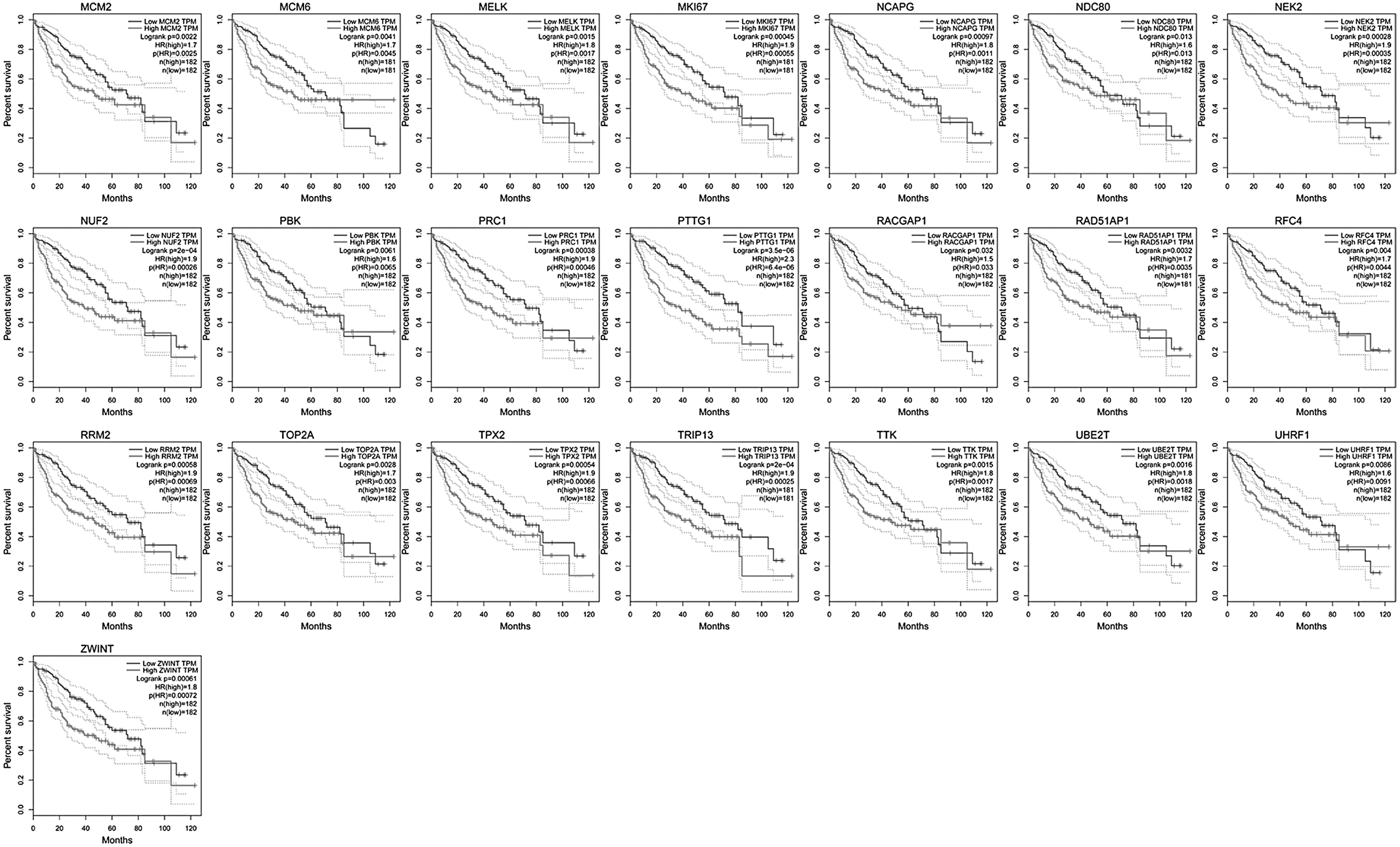
Overall survival analysis of prognostic biomarkers in accordance to TCGA/GTEx data sets. All genes are upregulated genes. Pictures can be found online. GTEx, Genotype-Tissue Expression; TCGA, The Cancer Genome Atlas.
Discussion
HCC is one of the world's most prevalent cancers. Its incidence is increasing and is closely related to advanced liver disease. Despite advances in medical and surgical treatments, HCC remains one of the most common causes of cancer-related deaths worldwide (Tang et al., 2017). Therefore, the search for new biomarkers is of great clinical importance to aid in early diagnosis and treatment decisions.
In summary, in this study, using in silico methods we identified candidate genes and signaling pathways associated with HCC. These genes and pathways were identified using a combination of DEG, GO, KEGG, and PPI-driven analyses. The results can serve as a starting point for the characterization and improved understanding of the underlying molecular pathogenic mechanisms leading to HCC. These identified candidate genes and pathways may also provide insights into new HCC therapeutic targets. Finally, we have discovered a new gene, DEP domain-containing protein 1B (DEPDC1B) that might serve as a biomarker for liver cancer.
DEPDC1B plays an important role in breast cancer and nonsmall cell lung cancer. Yi's team has demonstrated that DEPDC1B is significantly upregulated in nonsmall cell lung cancer cell lines and tissues (Yang et al., 2014). The high expression of DEPDC1B promotes the invasion and metastasis of nonsmall cell lung cancer lines. DEPDC1B-enhanced migration and invasion in non-small cell lung carcinoma is mediated through the Wnt/b-catenin pathway. Through querying the TCGA database, it was found that high expression of DEPDC1B was associated with poor patient prognosis (Yang et al., 2014).
Compared with normal prostate tissue, the expression level of DEPDC1B in tumor tissues of prostate cancer (PCa) patients was significantly increased. Increased expression of DEPDC1B was significantly associated with advanced clinical stage (p = 0.006), advanced T stage (p = 0.012), and lymph node metastasis (p = 0.004). Kaplan-Meier analysis showed that patients with high DEPDC1B mRNA levels had significantly shorter survival without biochemical recurrence (BCR). Therefore, the expression of DEPDC1B can be used as an independent predictor of survival time without BCR in PCa patients (Bai et al., 2017). Based on the earlier results and our bioinformatics analysis, we predicted that high expression of DEPDC1B in liver cancer is closely related to poor prognosis.
Other prognostic-related genes in liver cancer have been discovered and confirmed. For example, the relationship between the expression of the kinesin superfamily and the progression and prognosis of liver cancer has been reported to indicate that overexpression of KIF2C, KIF4A, and KIF11 is significantly correlated with lower survival of liver cancer (Chen et al., 2017). Upregulation of BUB1B, CDC7, and CDC20 in liver cancer tumor tissues predicts poor overall survival and disease-free survival in patients with HCC (Zhuang et al., 2018). In the liver cancer specimens with increased CECC-K expression, the DNA methylation status of the centromere protein K (CENP-K) promoter was significantly decreased. Overexpression of CENP-K stimulated tyrosine phosphorylation of AKT and MDM2 proteins, but inhibited TP53protein. Tyrosine phosphorylation, overexpression of the CENP-K gene in HCC promotes cell proliferation by activating AKT/TP53 signaling pathway (Wang et al., 2017). These results show that our research is consistent with the facts.
Overall, in this study, we identified 57 potential HCC-related candidate genes, most of which have previously been implicated in multiple pathways associated with tumorigenesis. Foremost, we identified a new prognostic-related gene DEPDC1B, as a potential marker for HCC, which has not previously been implicated in carcinogenesis. The results in this study might provide some directive significance for further exploring the potential biomarkers for diagnosis and prognosis prediction of HCC patients.
Footnotes
Authors' Contributions
Y.X.S. performed the comparative analysis using bioinformatics tools. Z.M.Z. interpreted the data and wrote the article. Both the authors participated in the data analysis, read and approved the final article.
Acknowledgment
We thank Professor Bin Zhao (Sheng Xin Zhu Shou) of Xiamen University for his guidance in this article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.