
Abstract
Aims:
Split-hand/split-foot malformation (SHFM) is a developmental and congenital limb malformation characterized by variable degrees of medial clefting or absence of one or more digits in hands and/or feet. The aim of this study was to identify the underlying cause of three consanguineous Pakistani families showing various types of SHFM-related features.
Materials and Methods:
Standard molecular methods, including whole-genome sequencing (WGS), whole-exome sequencing (WES), microsatellite markers-based genotyping, and Sanger sequencing were performed to search for the likely causative variants.
Results:
In family A, WES revealed a novel homozygous missense variant [c.338G>A, p.(Gly113Asp)] in the WNT10B gene. In family B, microsatellite-based genotyping followed by Sanger sequencing revealed a novel homozygous 13 base pairs deletion [c.884-896delTCCAGCCCCGTCT, p.(Phe295Cysfs*87)] in the same gene. In family C, WGS divulged a previously reported heterozygous missense variant [c.956G>A, p.(Arg319His)] in the TP63 gene.
Conclusions:
Mapping and sequencing genes and variants for severe skeletal disorders, such as SHRM, will facilitate establishing specific genotype-phenotype correlations and providing genetic counseling for the families suffering from such conditions.
Introduction
Split-hand/foot malformation (SHFM) is a congenital and developmental disorder of limb/digit primarily altering the central rays of hands and feet. It mostly occurs as a result of disruption of the Wnt-Fgf-Bmp molecular pathway. It is a classical form of ectrodactyly, also known as lobster claw malformation, which is often accompanied by hypoplasia of phalanx/syndactyly and aplasia of phalanges/metacarpals/metatarsals. Clinical spectrum of the SHFM differs from patient to patient in severity and even varies among individuals in the same family (Duijf et al., 2003; Elliott and Evans, 2008). It is inherited in autosomal dominant, autosomal recessive, and X-linked manner.
To date, 12 distinct types of SHFM and related phenotypes have been mapped on different human chromosomes. This includes eight various types of autosomal recessive and dominant SHFM (SHFM1-8) (Umair and Hayat, 2020), a locus mapped on chromosome 8q21.11-q22.3 (Gurnett et al., 2006) and three distinct types of split-hand/foot malformation with long-bone deficiency (SHFLD1-3). For eight various forms of SHFM, six causative genes, including DLX5 and DLX6 for SHFM1, TP63 for SHFM4, WNT10B for SHFM6, ZAK for SHFM7, and EPS15L1 for SHFM8, have been reported (Ugur and Tolun, 2008; Spielmann et al., 2016; Ullah et al., 2016, 2017; Umair et al., 2018; Umair and Hayat, 2020). SHFLD (split-hand/foot malformations with long-bone deficiency) inherited in dominant form is genetically different from isolated forms of SHFM. It is classified into three distinct types; SHFLD1 (MIM 119100) mapped on chromosome 1q42.2q43, SHFLD2 (MIM 610685) mapped on chromosome 6q14.1, and SHFLD3 (MIM 612576) mapped on chromosome 17p13.3. Microduplication involving BHLHA9 has been associated with SHFLD3 (Armour et al., 2011), however, no gene has been associated with SHFLD1 and SHFLD2. Furthermore, homozygous intragenic variants in the BHLHA9 gene also results in mesoaxial synostotic syndactyly associated with postaxial polydactyly (Malik et al., 2014; Ullah et al., 2019).
In the report, presented here, we have provided clinical and genetic characterization of three consanguineous families segregating SHFM either in autosomal recessive or autosomal dominant manner. Next-generation sequencing and microsatellite-based genotyping followed by Sanger sequencing revealed two novel homozygous variants in the WNT10B in two different families and a previously reported heterozygous variant in the TP63 in the third family.
Materials and Methods
Human subjects
Three families A-C, presented here, were recruited from different remote areas of Pakistan. Institutional Review Board (IRB) of Quaid-i-Azam University, Islamabad, Pakistan, approved to conduct the study. Informed written consent to conduct the study and publish the research data was obtained from all those who participated in the study.
Extraction of genomic DNA
Peripheral blood samples were collected in the EDTA containing vacutainer sets from 16 individuals, including two affected (V-1, V-2) and four unaffected (IV-1, V-3, V-4, V-5) members of family A
Whole-exome sequencing
Whole-exome sequencing (WES) using DNA of an affected member (V-1) of family A was performed on IlluminaHiSeq2500 platform using Agilent Sure Select Target Enrichment Kit as described previously (Umair et al., 2018). All the obtained reads were aligned using Burrows-Wheeler Aligner (BWA) and the variants were called using standard methods. Variant calling format file was generated and analyzed using Illumina BaseSpace online analysis tool. As the pedigree of family A depicted autosomal recessive inheritance pattern, therefore relevant filters (excluding synonymous, MAF >0.01, heterozygous variants) were applied accordingly. However, preference was given to homozygous variants.
Genotyping
In family B, search for the disease-causing gene was carried out by typing polymorphic microsatellite markers mapped in vicinity of the genes responsible for producing SHFM phenotypes. This included SHFM6 at chromosome 12q11-q13 (D12S361, D12S347, D12S1677, D12S297, D12S368, D12S270, and D12S1604). Analysis of the PCR-amplified markers, haplotype construction, purification of the products, and further analysis were the same as described previously (Ullah et al., 2018).
Whole-genome sequencing
DNA of two affected members (VI-1, IV-2) of family C was subjected to whole-genome sequencing (WGS) at Novogene (Sacramento, CA). Sequencing library was prepared using TruSeq Nano DNA HT Sample Preparation Kit (Illumina, CA) following manufacturer's instructions. Sequencing was performed on Illumina HiSeq X system (HiSeq PE150, Q30 ≥ 80%) to an average depth of 9X. Genome sequencing data were aligned, annotated, and filtered using Genoox pipeline. Segregation of the variant was analyzed by performing Sanger sequencing using DNA of rest of the affected and unaffected members of the family C.
Sequencing WNT10B and TP63
Primers, to PCR-amplify exons and splice junctions of the two genes, WNT10B and TP63 were designed using online primer3 tool. PCR amplified products were purified with a commercially available kit (Axygen, CA). Sanger sequencing was performed bidirectionally using standard protocol as described previously (Umair et al., 2016).
In silico analysis
Primary protein sequence of the WNT10B was retrieved through Ensemble genome browser and subjected to BLAST search against PDB (Protein Data Bank) database for search for a suitable template. We used Xenopus leavis WNT8 structure (PDB ID: 4F0A) as template for homology modeling approach. Subsequently, Ramachandran plot and ERRAT (Colovos and Yeates, 1993) tools were utilized to validate the predicted three-dimensional (3D) models followed by its refinements and geometry optimizations using WinCoot (Emsley et al., 2010), UCSF Chimera 1.7.0 (Meng et al., 2006), and VEGA ZZ tools.
Modeled protein structures were docked against Frizzled-8 (PDBID: 4F0A) using PatchDock (Morris et al., 2009).
Results
Clinical features
Family A
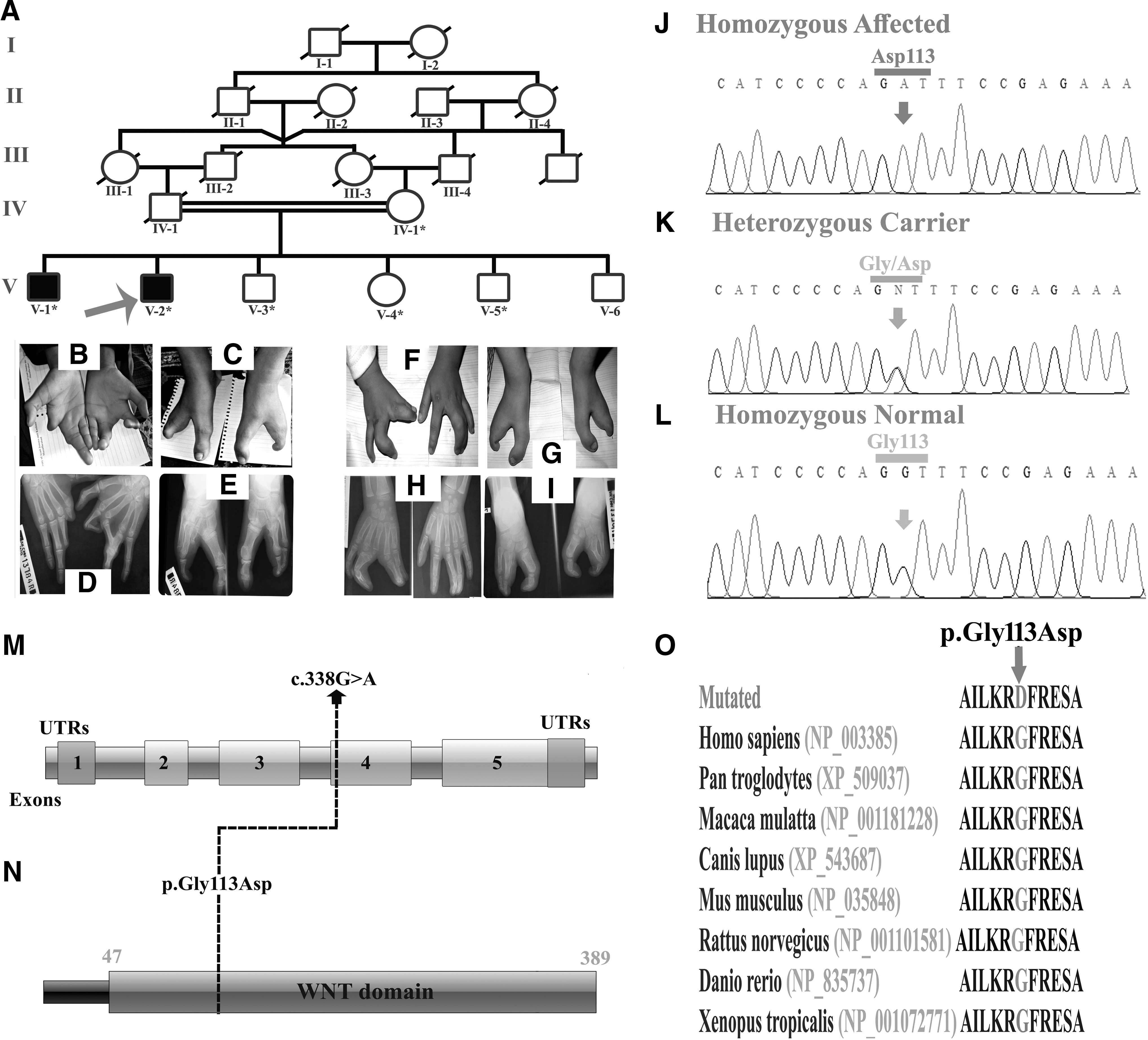
In this family, two affected individuals (V-1; 19 years) and (V-2; 17 years) displayed severe form of SHFM. The elder affected individual (V-1) had severe limb malformation such as central type cleft hand. The right hand showed agenesis of metacarpal bones, including missing 2nd and 3rd digit. The left hand showed dysplastic cleft associated with 1st finger camptodactyly and hypoplasia of 2nd finger. Feet showed association of cleft with syndactyly. Radiographic examination revealed incomplete development of the digits and missing nails in the feet (Fig. 1B-E). Long-bone anomaly was not observed on skeletal radiographs. The younger affected member (V-2) showed syndactyly of middle and ring fingers and mild camptodactyly of middle finger in the left hand. The right hand showed fusion of thumb and index finger, and mild central median cleft (Fig. 1F-I). Features in the feet were similar to those observed in affected member (V-1). Heterozygous carriers did not show any type of bone malformation.
Family B
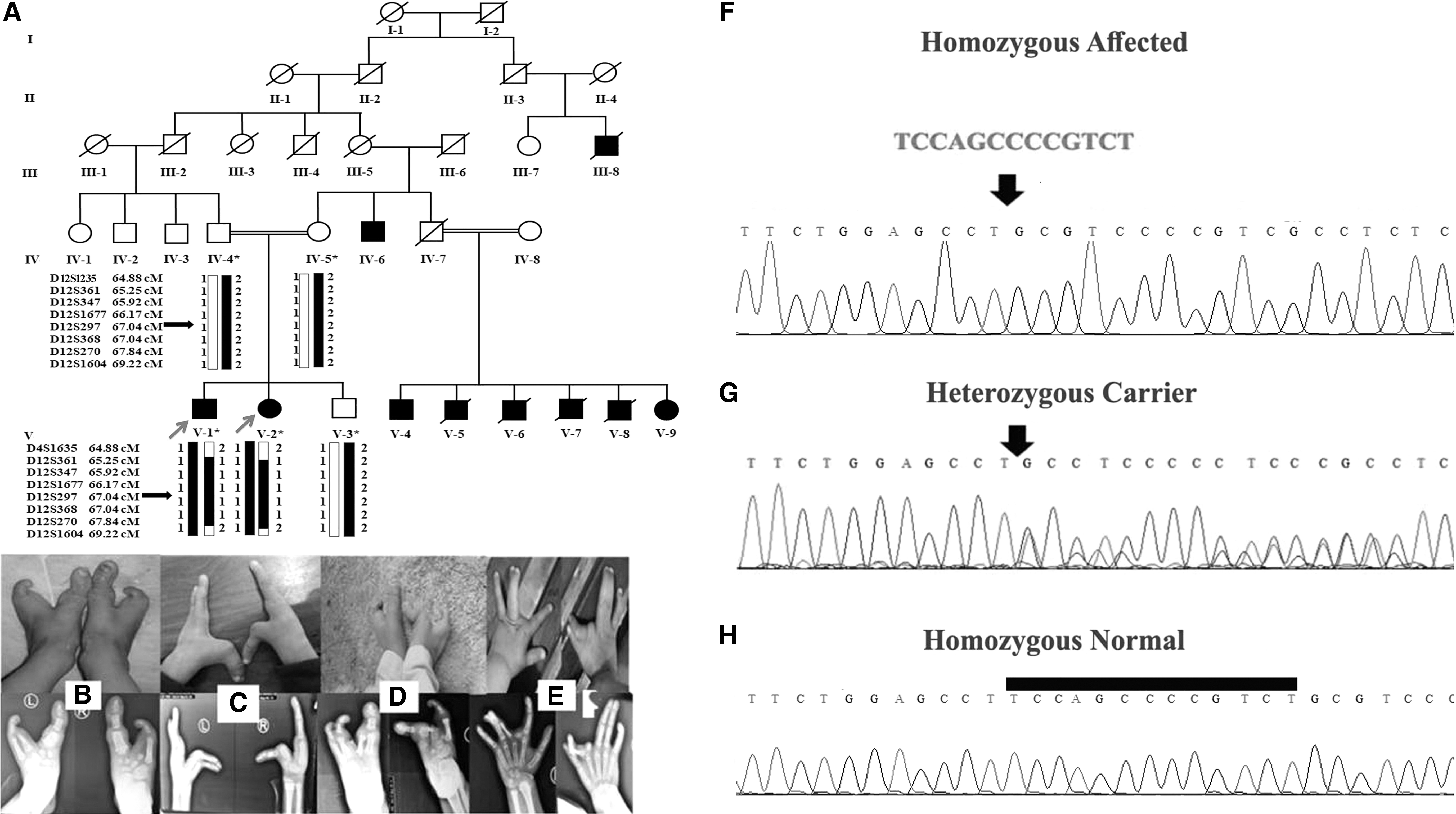
Affected individuals in family B showed classical phenotypes of SHFM. At time of the study, age of the patients (V-1) and (V-2) were 6 and 4 years, respectively. They were born with full-term pregnancies without any complication. Radiographic analysis of the patient (V-1) showed cleft hand malformation with central type syndactyly involving 3rd and 4th fingers in the left hand. The middle finger in the left hand and 2nd, 3rd, and 4th fingers in the right hand were missing. In the same affected member 2nd, 3rd, and 4th toes in the right foot, and 2nd and 3rd toes and distal phalange on 4th toe in the left foot were missing (Fig. 2B, C). Radiographs of the second affected individual (V-2) showed cleft hand malformation associated with camptodactyly in the right index finger and clinodactyly in the right small finger. In addition, syndactyly involving 3rd and 4th fingers was observed on both hands (Fig. 2D, E).
Family C
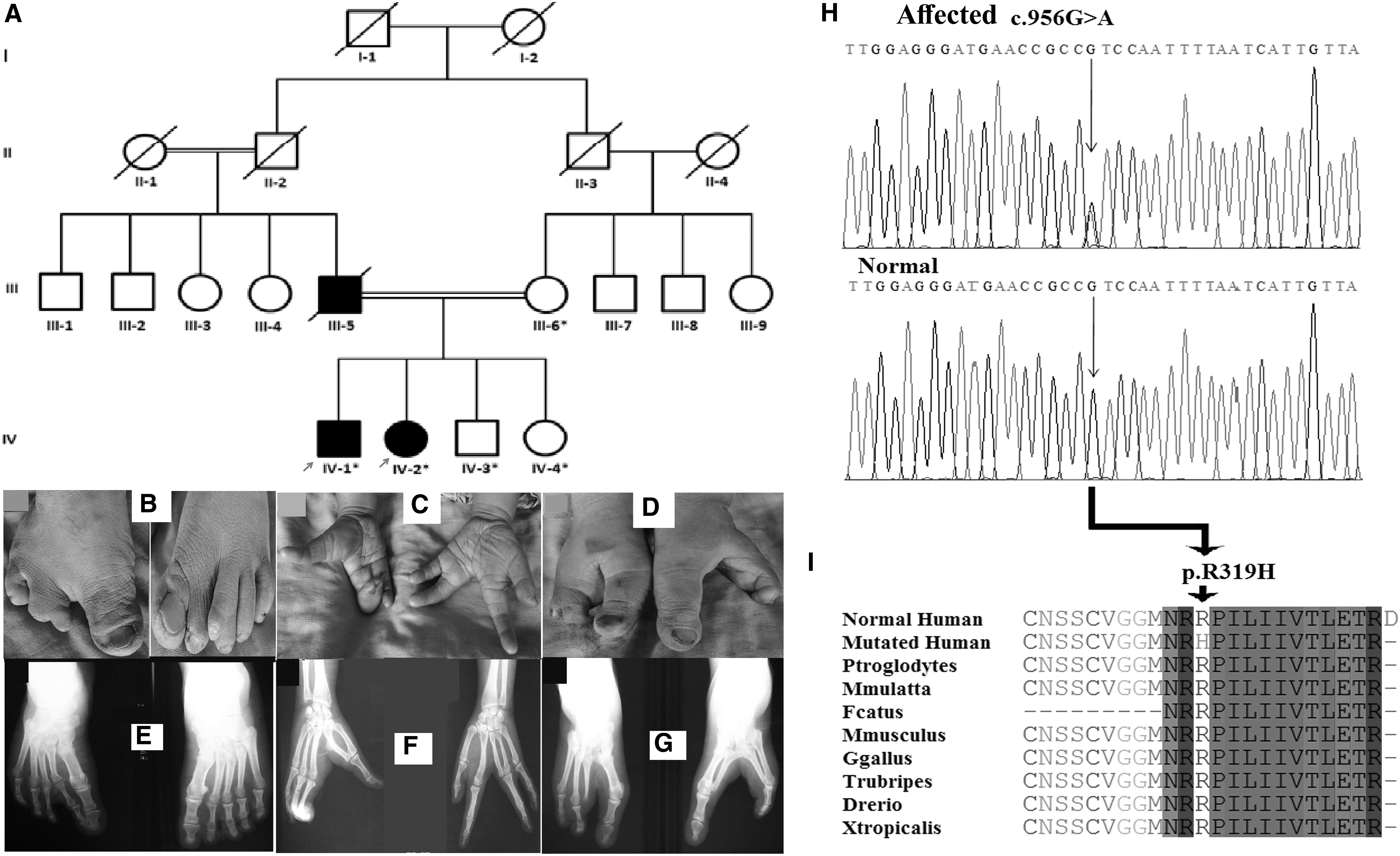
In this family, affected individual (IV-1) showed fusion of great toe and 2nd toe, and duplicated 3rd toe in the left foot. In the right foot, great toe and 2nd toe were fused to develop into a condition called oligodactyly. Hands were apparently unaffected. Another affected individual (IV-2) in the same family showed both cleft feet and hands. The cleft feet resulted from missing central toes 2-4 in the right and 2-3 in the left foot. Similarly, hands showed aplasia of 2nd finger in the right hand and 3rd finger in the left hand (Fig. 3A-G). The matriarch verified that her husband (III-5) had SHFM-typed limbs validating segregation of the disorder in autosomal dominant manner in the family.
Molecular analysis
Family A
Disease causing gene in this family was carried out by subjecting DNA of an affected member for WES. Screening to select the candidate variants was based on inheritance of the disorder in autosomal recessive pattern and clinically presenting SHFM phenotypes. Total 181 homozygous variants were selected after applying different filters (NHLBI-ESP; 1000 Genomes; ExAC with MAF <0.01). Based on previous published reports (Ugur and Tolun, 2008; Khan et al., 2012, 2020; Aziz et al., 2014; Ullah et al., 2018) describing involvement of the WNT10B in causing SHFM, a variant (c.338G>A) (NM_003394.3; Human GrCh38) in the same gene was selected for further analysis. This led to the identification of a novel homozygous missense variant p.(Gly113Asp) in exon 4 of the WNT10B (MIM 601906). Bidirectional Sanger sequencing of DNA of rest of the affected and unaffected members validated segregation of the variant with the disease phenotype within the family (Fig. 1J-L).
Family B
Analysis of haplotype, constructed based on typing microsatellite markers, established linkage in the family on chromosome 12p11 harboring the WNT10B (Fig. 1A). Bidirectional sequencing of the gene identified a novel homozygous 13 base pairs gross deletion [c.884-896delTCCAGCCCCGTCT, p.(Phe295Cysfs*87)] in exon 5 of the gene (Fig. 2F-H). Sequencing rest of the family members validated segregation of the variant with the disease within the family.
Family C
Due to consanguinity among parents of affected individuals in two generations of family C, the pedigree was considered to segregate disease phenotypes in autosomal recessive form. Microsatellite markers-based homozygosity mapping was performed for previously SHFM-associated genes. No homozygosity shared by all affected individuals was found using microsatellite markers closely linked to the SHFM-associated genes. To search for the disease-causing variant in the family, WGS was performed on DNA of two affected members (IV-1, IV-2). After excluding synonymous variants, the variants shared by both the affected individuals were selected for further analysis. In total, 113 heterozygous variants were found common in both affected individuals. Based on involvement of a previously reported TP63 in causing SHFM, a heterozygous variant (c.956G>A) in the same gene was selected for further analysis. Sanger sequencing of DNA of rest of the affected and unaffected members validated segregation of the missense variant [c.956G>A p.(Arg319His)] with the disorder within the family (Fig. 3H-I).
Pathogenicity index
All three variants, including two novel and one previously reported were declared pathogenic using different online prediction tools such as Mutation Taster, DANN, FATHMM-MKL,SIFT, and PROVEAN. Analysis by HomoloGene-NCBI revealed that the mutated amino acids were highly conserved across different species (Fig. 1O). The variants identified in family A and B were not found in homozygous state in dbSNP, 1000 Genomes, exome variant server, internal database, ExAC, and gnomAD. Both variants were screened in 155 ethnically matched controls and 145 in house exomes.
Structural analysis
Structural analysis of a missense variant p.(Gly113Asp), identified in the WNT10B in family A, was performed by generating 3D models of wild-type and mutated WNT10B using homology modeling. It was then characterized by online available structural analysis tools. Ramachandran scores of WNT10bWT and WNT10bGly113Asp (98.98%) and FZD8 (100%) suggested that majority of amino acids were lying in the sterically allowed regions on the plot. PatchDock predicted energy values for WNT10bWT and WNT10bGly113Asp in association with FZD8 were −25.5 Kcal/mol and −27.7 Kcal/mol, respectively. In case of WNT10bWT and FZD8 complex, three residues Arg88, Arg360, and Gln358 in WNT10bWTwere involved in interaction with Glu77, Val76, and Gln79 in FZD8. However, in case of WNT10bGly113Asp and FZD8 complex, four other residues Pro37, Pro41, Arg233, and Pro301 in WNT10bGly113Asp were involved in interaction with four different residues, including Pro41, Cys70, Gln97, and Lys120 in FZD8. There was obvious difference in binding of FZD8 with WNT10bWTand WNT10bGly113Asp. This is due to backward movement of β3 and β4 in the WNT10bGly113Asp, which in turn disrupted binding region leading to variation in protein function (Supplementary Fig. S1A-C).
For variant p.(Phe295Cysfs*87), identified in the WNT10B in family B, Ramachandran plots for WNT10BWT and WNT10BPhe295Cysfs*87 indicated >93% of residues in allowed region (Supplementary Fig. S2A, B). Structural analysis revealed completely altered overall topology of the protein, including disruption in intrastructural disulfide bridges and helix-helix interactions (Supplementary Fig. S2C-H). It was verified by molecular docking of WNT10BWT and WNT10BPhe295Cysfs*87 against FZD8. The WNT10BWT showed hydrogen bonding with FZD8 through His85, Arg88, Glu4, Ser12, Cys366, and Arg360 (Supplementary Fig. S2I) and hydrophobic interaction through Leu20, Leu340, Asn335, Ser338, Thr337, Arg339, Leu341, Gly16, Pro7, Val381, Trp370, His369, Phe368, Val374, Cys376, Arg363, Cys364, His365, Arg367, Glu362, Cys379, Thr382, Glu383, Trp384, Val385, and Leu2 (Supplementary Fig. S2K). However, WNT10BPhe295Cysfs*87 showed hydrogen bonding with FZD8 through Cys23 and His77 (Supplementary Fig. S2J) and hydrophobic interactions through Ser358, Gly380, Ile361, Ala362, Ala363, Gln84, His81, Arg88, His85, Pro5, Arg269, Leu22, Trp337, Gln74, Arg6, Val340, Glu4, Glu3, Ser374, Leu97, Thr365, Ala375, Met373, Arg102, Gly100, Leu73, Glu98, Pro10, Arg8, Pro9, Pro7, Ala359, Leu357, and Glu356 (Supplementary Fig. S2L).
Discussion
In the present investigation, we have clinically and genetically characterized three families representing various features of SHFM. Families A and B segregated the disorder in autosomal recessive and family C in autosomal dominant manner.
Affected members in all the three families showed combinations of previously reported features of SHMF, including cleft hand/foot malformation, radial ray malformation, polydactyly, syndactyly, hallux valgus malformation, aplasia, hypoplasia, hypoplastic finger, and missing of phalanges (Ugur and Tolun, 2008; Khan et al., 2012; Aziz et al., 2014; Ullah et al., 2018). Features associated with disease causing variants in the WNT10B, including oligodontia/dental anomalies (Yu et al., 2016; Kantaputra et al., 2018) and obesity (Christodoulides et al., 2006), were not observed in any affected member of the three families presented here.
In two families, A and B, disease causing variants were identified in the WNT10B and in family C in the TP63. This included a novel missense variant p.(Gly113Asp) in family A and a novel frameshift variant p.(Phe295Cysfs*87) in family B in the WNT10B, and a previously reported missense variant p.(Arg319His) in family C in the TP63. To date, only 20 mutations have been reported in the WNT10B causing SHFM phenotypes (Human Gene Mutation Database). Four of these have been reported in Pakistani population. This included a homozygous missense variant p.(Thr329Arg) (Khan et al., 2012), 4bp deletion (c.1165_1168delAAGT), 7bp duplication (c.300_306dupAGGGCGG) (Aziz et al., 2014), and a homozygous nonsense variant p.(Gln154*) (Ullah et al., 2018).
The WNT10B contains five exons, spans 6.42 Kb on chromosome 12q11-q13, and encodes 389 amino acids protein (UniProtKB/Swiss-Prot accession 000744). The WNT10B is one of the members of WNT gene family, which contains at least 19 other genes. WNT proteins are known to play role in fetal limb bud growth, development, and adult hematopoiesis by activation of the canonical signaling pathway by acting as ligands for cell surface receptors complexes composed of frizzled (FZ) and low-density lipoprotein receptor-related protein 5/6 (LRP5/6) family members (Gordon and Nusse, 2006). The WNT10B expression has been reported in various stem cell compartments, including postnatal growth plate, bone marrow, and osteoblastic precursors (Andrade et al., 2007; Zhou et al., 2008; Brunelle et al., 2019). The study of null mice exhibited loss of Wnt10b that results in 30% reduction volume and mineral density of the bone (Bennett et al., 2006). Stevens et al. (2010) have shown that maintenance of adult bone density is dependent on presence of both alleles of the WNT10B.
The missense variant p.(Gly113Asp), located in conserved WNT domain in the WNT10B (Supplementary Fig. S3), is likely to disrupt the protein binding to its receptor FZD8 as shown by the molecular docking studies. In case of the frameshift variant p.(Phe295Cysfs*87), molecular docking approaches showed deletion of the amino acids at critical position affected the overall topology of the WNT10B by causing significant structural twists that influence the formation of intrastructural disulfide bridges and helix-helix interactions with the receptor FZD8. Therefore, deep structural analysis of the two identified variants in the WNT10B marks its implication in SHFM pathogenesis. In addition, bioinformatic analysis also found both the variants damaging and disease causing.
As reported previously, the missense variant p.(Arg319His) in the TP63, identified in the family C, can affect its binding to DNA. Arginine at position 319 is likely to be involved in maintenance of the overall structure of the DNA-binding domain. This variant was previously reported by Van Bokhoven et al. (2001) in a family of European origin presenting features of Ectrodactyly-ectodermal dysplasia cleft lip/palate (EEC). In addition to SHFM, the author reported additional phenotypes of sparse hair, lacrimal-duct abnormalities, nail dystrophy, dry skin, and anodontia/hypodontia in the affected individuals. Affected individuals in family C did not show any abnormality other than ectrodactyly, thus showing segregation of nonsyndromic SHFM. An affected individual (IV-1) in family C showed polydactyly phenotype that has not yet been reported in SHFM patients harboring variants in TP63. Variation in phenotypes caused by the same mutation may be due to the fact that patients have different familial backgrounds. To date, 10 heterozygous variants, including seven missense, one splice site, and two nonsense, have been reported in the TP63. The missense variant p.(Arg319His) in the TP63, identified in the family C, is the first familial case of SHFM4 in Pakistani population.
In conclusion, we have reported three disease-causing variants in three families, including two novels in WNT10B and one in TP63, resulting in SHFM phenotypes. The absence of ectodermal phenotypes in present patients segregating previously reported variant in TP63 highlights the importance of modifier genes causing variations in TP63-related SHFM phenotypes.
Footnotes
Declaration
The data that support the findings of this study are available on request from the corresponding author.
Acknowledgments
We highly appreciate participation of the family members in the study. M.B. was supported by indigenous PhD fellowship from HEC, Islamabad, Pakistan.
Ethics Statement
Written informed consent for the presentation and publication of this study was obtained from all the participating members.
Authors' Contributions
M.B., A.H., M.U., and A.U. performed laboratory experiments and drafted the article. N.B., S.K., and E.M. performed protein analysis. S.B. performed exome sequencing. M.I.M. collected the samples. Umm-e-Kalsoom, W.A., and B.K. designed the study, finalized the article, and provided funds for the study presented.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.