
Abstract
Background:
Muscular dystrophies are a heterogeneous group of inherited disorders that cannot be diagnosed clinically due to overlapping clinical phenotypes. Whole-exome sequencing is considered as the diagnostic strategy of choice in these cases. In this study we aimed to determine the mutational spectrum of multiplex ligation-dependent probe amplification (MLPA)-negative muscular dystrophy patients in Pakistan using whole-exome sequencing. Subsequently the mutations identified via WES were used to screen additional dystrophinopathy patients by Sanger sequencing.
Materials and Methods:
DNA extracted from the peripheral blood of three MLPA-negative muscular dystrophy patients was sent for whole-exome sequencing. The identified variants in these 3 patients were then checked in 18 dystrophinopathy patients using Sanger sequencing.
Results:
Four missense variants and one nonsense variant in the Duchenne muscular dystrophy (DMD) gene were detected. WES diagnosed a DMD patient carrying a nonsense variant c.4375C>T (rs398123953) who can benefit from Ataluren therapy. The other two patients carried missense variant (c.572G>T) in the YARS2 gene (rs11539445) labeling them as patients of MLASA (myopathy, lactic acidosis, and sideroblastic anemia). The identified missense and nonsense variants in the DMD gene were detected in 18 clinically diagnosed dystrophinopathy patients using Sanger sequencing. Three missense variants were detected in our cohort of 18 dystrophinopathy patients. One missense variant c.3406A>T (rs3827462) and a nonsense variant c.4375C>T (rs398123953) were not detected in our cohort of 18 dystrophinopathy patients.
Conclusions:
Whole-exome sequencing identified a nonsense variant in Pakistani muscular dystrophy patients, which is amenable to treatment by Ataluren and a missense variant in YARS2 gene responsible for causing MLASA.
Introduction
Muscular dystrophies are a heterogeneous group of muscular disorders particularly affecting voluntary muscles. They are classified according to their mode of inheritance and pattern of muscle involvement. The various types include congenital muscular dystrophy, myotonic dystrophy, Becker muscular dystrophy (BMD), Duchenne muscular dystrophy (DMD), facioscapulohumeral dystrophy, Emery-Dreifuss muscular dystrophy, oculopharyngeal muscular dystrophy, distal muscular dystrophy, and limb girdle muscular dystrophy (Emery, 2002).
Dystrophinopathies, as suggested by the name, include disorders due to mutation in the dystrophin gene affecting the production of dystrophin protein. Dystrophinopathies have been classified into X-linked dilated cardiomyopathy (XL-DCM), BMD, and the most severe, DMD (Govoni et al., 2013; Toksoy et al., 2019; Tomar et al., 2019).
Dystrophin protein is a member of the dystrophin glycoprotein complex, which maintains cellular architecture and assists in mobility (Straub and Campbell, 1997). DMD is a fatal condition with an incidence of 1 in 3500 live male births and occurs due to mutation in the dystrophin gene causing lack of dystrophin protein production (Emery, 1991, 2002). BMD is a comparatively milder form of dystrophinopathy with an incidence of 1/18,500 live male births (Bushby et al., 1991). The mutation in BMD does not disrupt the reading frame, resulting in partially functional dystrophin protein production (Govoni et al., 2013). XL-DCM is associated with ventricular enlargement and decreased ejection fraction, leading to heart failure. The dystrophin protein deficiency in this case is limited to the involvement of cardiac muscles, whereas skeletal muscle integrity and functioning are conserved (Govoni et al., 2013).
Large deletions account for most of the identified cases (80%) and the remaining 20% are small mutations. Of these 52% of cases have point mutations, 25% deletions, 9% duplications, 14% splice site mutations, and in 0.3% of cases mid-intronic mutations have been observed (Bladen et al., 2015).
If large mutations are not detected through multiplex ligation-dependent probe amplification (MLPA), the diagnostic strategy must proceed to the detection of small mutations by sequencing individual exons (Aartsma-Rus et al., 2016). Since it is a laborious procedure to amplify each exon by polymerase chain reaction (PCR) and then sequence it by Sanger sequencing, the next-generation sequencing (NGS) can serve as a single platform for detecting most mutations simultaneously. Thus, it is considered an efficient and time-saving strategy (Deepha et al., 2017).
Two therapeutic strategies have been approved for dystrophinopathies. Eteplirsen involves skipping of exon 51, and Ataluren is a premature stop codon read through therapy, which is specifically designed for patients with nonsense mutation (Haas et al., 2015; Aartsma-Rus and Krieg, 2017). Recently, Golodirsen, which involves skipping of exon 53, has been proposed for treatment of DMD (Aartsma-Rus and Corey, 2020).
Our study aimed to detect small mutation in MLPA-negative dystrophinopathy patients using whole-exome sequencing. The purpose was to establish precise genetic diagnosis and identify candidates who can benefit from Ataluren treatment. The variants identified by whole-exome sequencing were assessed in other dystrophinopathy patients by Sanger sequencing to determine the most prevalent small mutations in our cohort.
Materials and Methods
Whole-exome sequencing
Three clinically diagnosed muscular dystrophy patients with negative MLPA results were selected for whole-exome sequencing based upon the clinical features listed in Supplementary Table S1.
The study was approved by institution's review board (IRB-771/DUHS/Approval/2016/293), and informed consent was obtained from all candidates included in the study.
DNA extraction was performed using AllPrep DNA/RNA Kit (Qiagen). DNA quantification was performed using NanoDrop (Thermo Fisher Scientific), and samples were sent for whole-exome sequencing to a commercial sequencing service. Whole-exome sequencing was performed on an Illumina platform, and Agilent SureSelect V6 post capture kit was used. Using the Burrows-Wheeler Alignment Tool (BWA), the FASTQ files were aligned to human reference genome 19 from University of California, Santa Cruz. Analysis was conducted using Picard and Genome Analysis Toolkit. For variant annotation, SnpEff (SnpEff_v4.1g), dbSNP database (version 142), 1000 Genome Phase3, ClinVar database (version 05/2015), and ESP database (ESP6500SI_V2) were used as shown in Supplementary Figure S1.
Selection of variants and in silico analysis of their impact
The variants in various genes were selected based on the following criteria:
Variants predicted to have a functional impact on protein coding regions of genes (missense variants, nonsense variants). Variants determined as damaging or probably damaging by in silico predictive mutation impact software SIFT and PolyPhen-2. Variants with clinical significance determined as likely pathogenic and pathogenic in the ClinVar database. Variants associated with various diseases in the NCBI database and Ensemble database.
Sanger sequencing
Sanger sequencing of respective amplified variants detected by whole-exome sequencing data was performed in 18 dystrophinopathy patients based upon the clinical features listed in Supplementary Table S2. One hundred nanograms of DNA was amplified using 0.25 μM primers and 2x Master Mix. (Primer sequences listed in Supplementary Table S3).
The PCR cycling conditions were as follows. Initial denaturation at 94°C for 3 min, followed by 30 cycles of denaturation at 95°C/15 s, annealing at 50.5°C/20 s, and extension at 72°C/30 s. Final extension was performed at 72°C for 7 min.
Three microliters of the amplified PCR product was resolved on 1.5% agarose gel and visualized under UV transilluminator. The remaining amplified PCR product was ethanol precipitated and resuspended in 21 μL TE buffer. The prepared samples were sent for Sanger sequencing to a commercial sequencing service. The forward primer of each reaction was used for sequencing. The results of Sanger sequencing were analyzed by comparison with the GenBank sequence of the DMD gene (NM_004006.2) (Variants analyzed through Sanger sequencing listed in Supplementary Table S4). The sequencing data were analyzed using SnapGene Viewer 4.1.5.
Results
Molecular diagnosis by whole-exome sequencing
Our strategy to perform whole-exome sequencing detected molecular alterations in all the three patients with atypical clinical features of muscular dystrophy. In total, six molecular alterations were detected in the three patients. Of these, four were missense variants in the DMD gene and one was a nonsense variant in the DMD gene. A missense variant in the YARS2 gene was detected in two of our patients, as shown in Figure 1 (Supplementary data S1-S2S3).
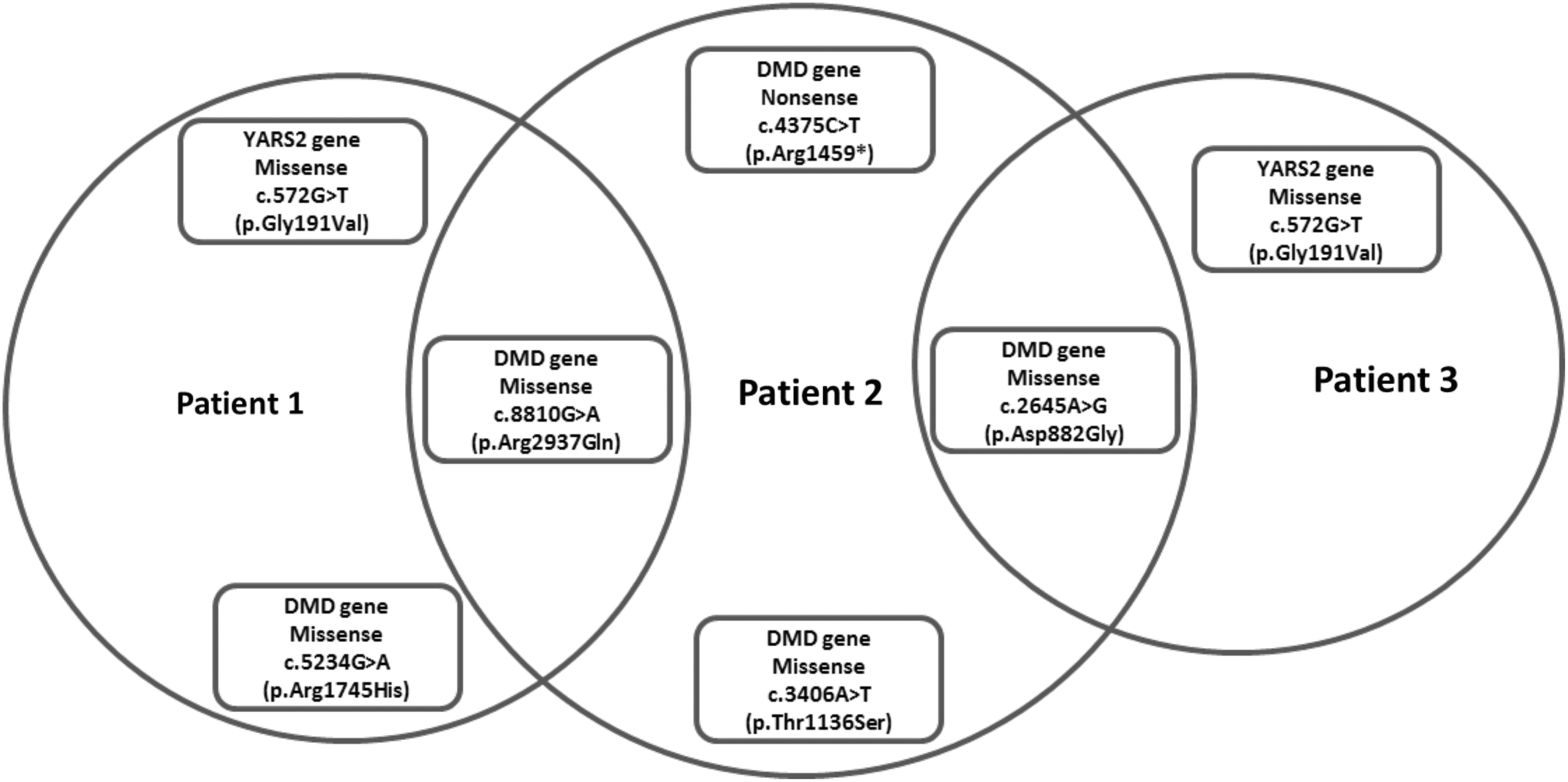
Variants detected by whole-exome sequencing in three patients.
Two of the missense variants were common in two patients. The missense variant c.8810G>A involving exon 59 of the DMD gene and the missense variant c.2645A>G involving exon 21 of the DMD gene were detected in two patients. The other missense variants were c.5234G>A involving exon 37 of the DMD gene and c.3406A>T involving exon 25 of the DMD gene.
A nonsense variant c.4375C>T involving exon 32 of the DMD gene was detected in one patient with a strong family history of muscular dystrophy. The whole-exome sequencing results concluded that the patient has DMD after clinical correlation.
All five molecular alterations have been reported in the Leiden open variant database (LOVD). In silico analysis was performed for the detected mutations using predictive mutation impact software SIFT and PolyPhen-2. Most of the identified variants were classified as benign and tolerated, whereas only two of them were classified as damaging and pathogenic by both the software. Nonsense variant c.4375C>T involving exon 32 of the DMD gene was described as pathogenic by the ClinVar database. c.5234G>A involving exon 37 of the DMD gene was predicted as damaging by SIFT and probably damaging by PolyPhen-2.
A missense variant c.572G>T involving exon 1 of the YARS2 gene has been detected in two of the patients screened by whole-exome sequencing. The variant has been reported in the LOVD and has been described as pathogenic by the ClinVar database. Both the patients carrying c.572G>T in the YARS2 gene have been diagnosed as cases of MLASA (myopathy, lactic acidosis, and sideroblastic anemia) based on the information provided in the Ensemble database and genotype-phenotype correlation. Table 1 summarizes the detailed features of each variant detected by whole-exome sequencing in three patients.
Analysis of Whole-Exome Sequencing Result of Muscular Dystrophy Patients Having Atypical Clinical Features
DMD, Duchenne muscular dystrophy; LOVD, Leiden open variant database; NAF, not affect function.
Sanger sequencing results
The dystrophin gene, due to its size, has many reported polymorphisms. Five single nucleotide polymorphisms of DMD gene were analyzed (rs228406, rs398123953, rs3827462, rs1800280, and rs1801187) using Sanger sequencing. Figure 2 shows the gel electrophoresis images of amplified PCR products.
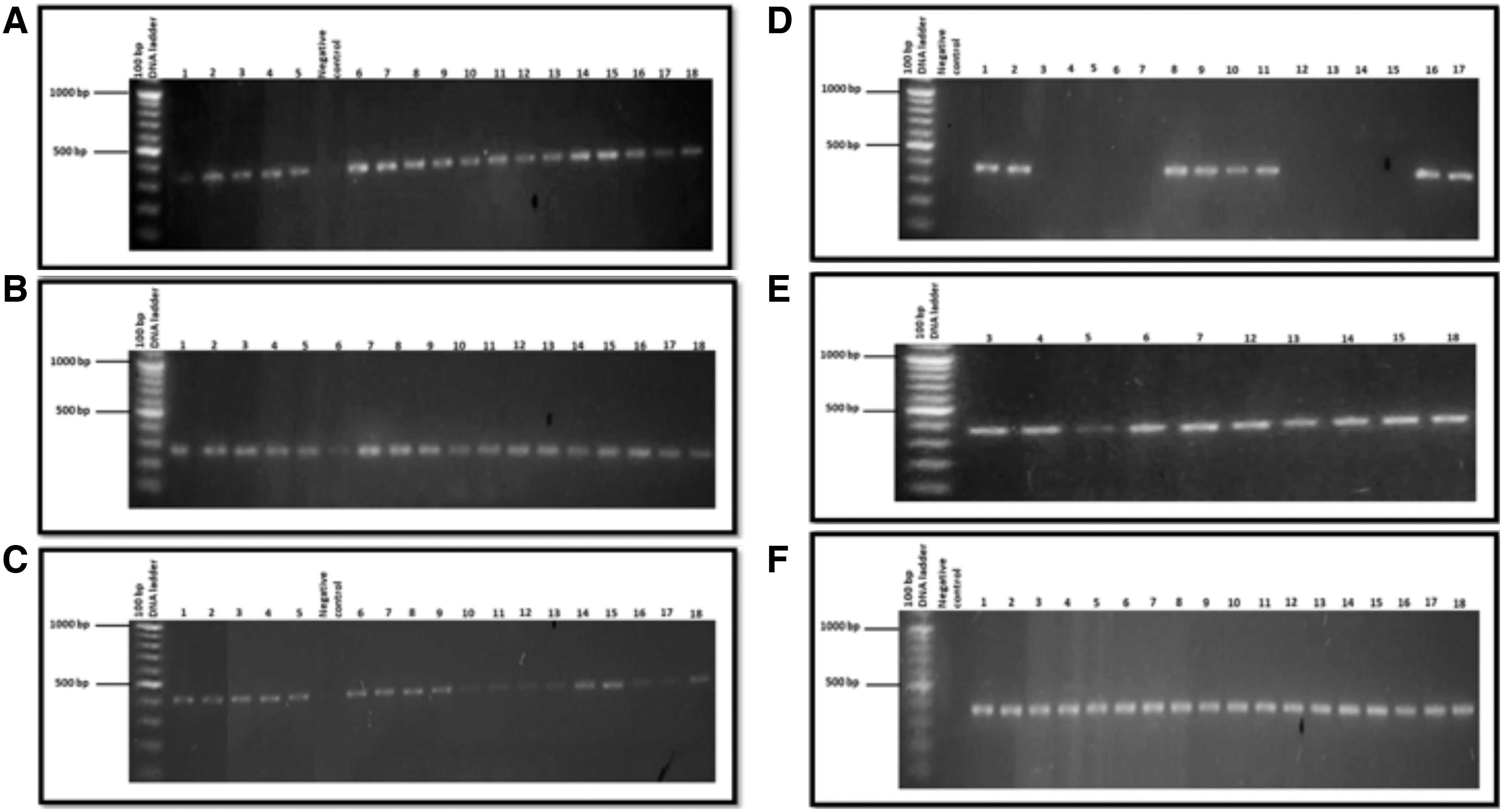
Gel electrophoresis images of amplified PCR products using primers complimentary to rs228406, rs398123953, rs3827462, rs1800280, and rs1801187 amplifying specific regions within the exons of DMD gene. GeneRuler 100 bp DNA ladder was used as a molecular size marker in the first lane. Patients were abbreviated as 1-18, and patient's samples were loaded in the subsequent lanes. No band is seen in the lane labeled as negative control
Description of variants on electropherogram
The sequencing data are generated in the form of four colored electropherograms. Each nucleotide is represented by peaks of different colors. In case of a heterozygous variant, the peaks had twin colors and overlapped each other. The presence of a homozygous variant is shown by the presence of single colored peak for that particular nucleotide. Figure 3 shows the variants detected on electropherogram.
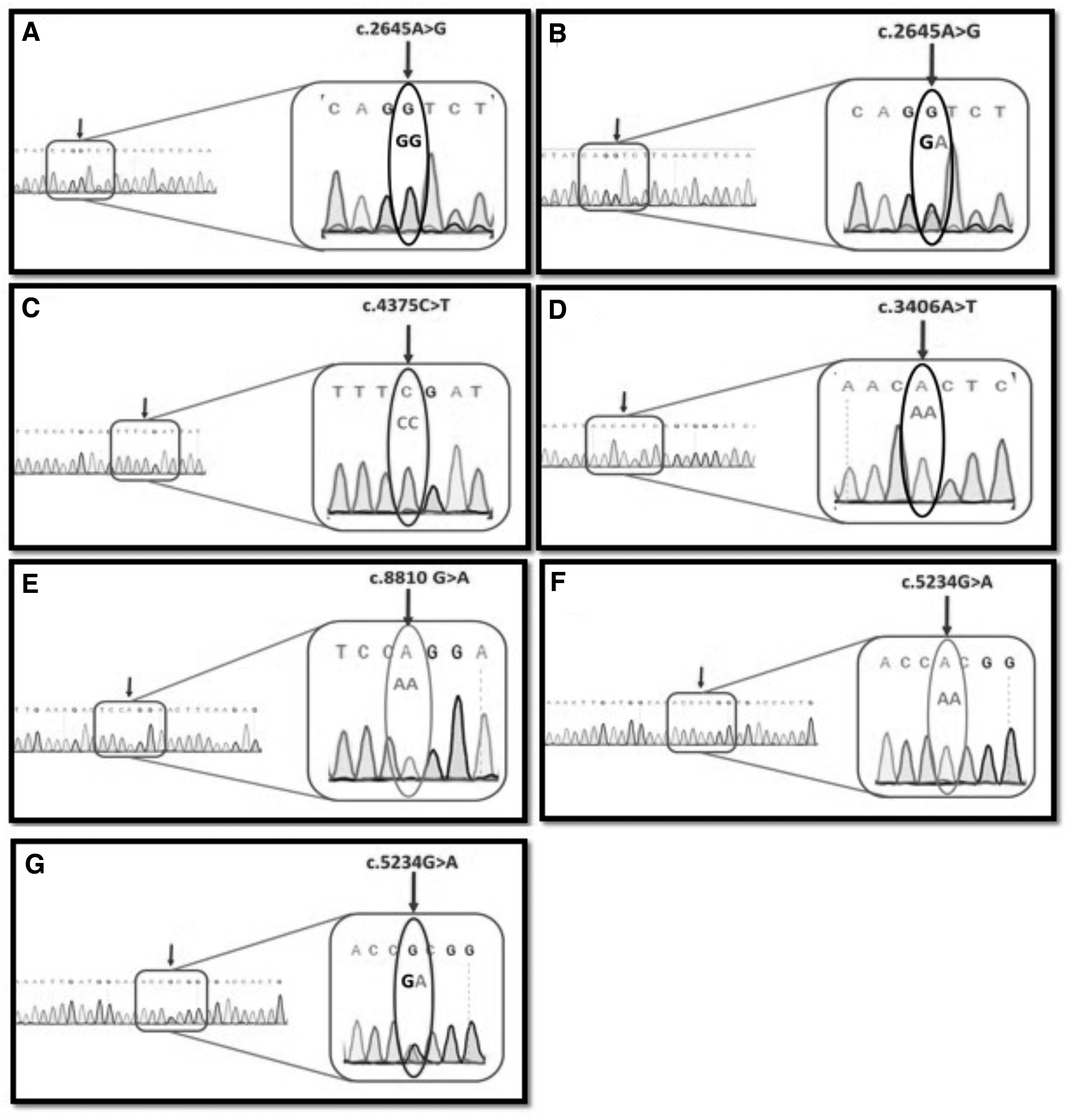
Detection of variants on electropherograms.
Mutational spectrum of the 18 patients screened by Sanger sequencing
c.2645A>G (rs228406) in exon 21 of the DMD gene was detected in all the 18 patients by Sanger sequencing. c.8810G>A (rs1800280) involving exon 59 of the DMD gene has been detected in 66.60% of patients and c.5234G>A (rs1801187) involving exon 37 of DMD gene was detected in 72.20% of patients screened by Sanger sequencing.
c.4375C>T (rs398123953) and c.3406A>T (rs3827462) were not detected in any of the 18 patients screened by Sanger sequencing. Figure 4 shows the mutational spectrum of the 18 dystrophinopathy patients screened by Sanger sequencing as mentioned in Supplementary Table S5.
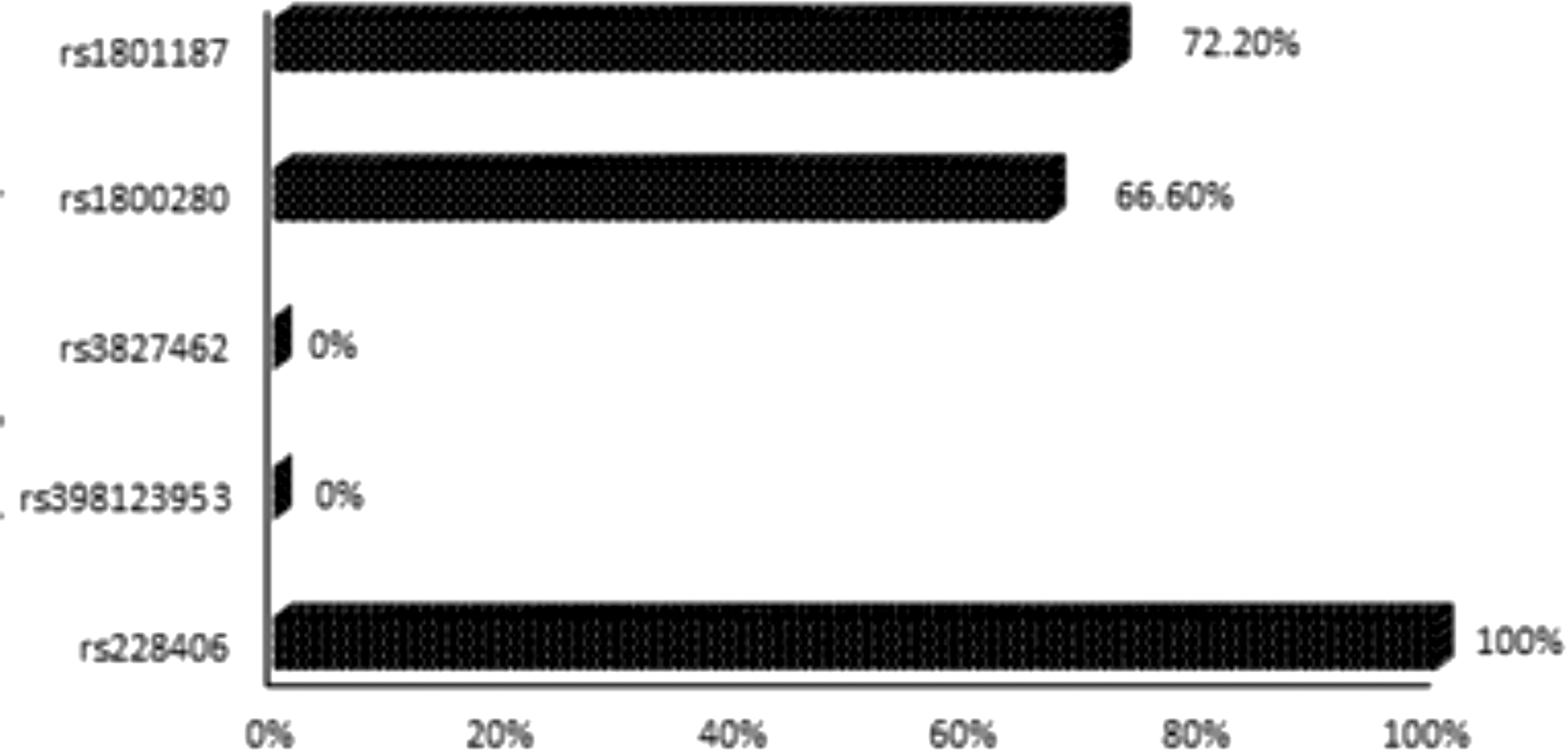
Mutational spectrum of 18 dystrophinopathy patients screened by Sanger sequencing.
Combination patterns of variants in study subjects
61.11% of all the 18 patients screened by Sanger sequencing certainly had c.2645A>G (rs228406), c.8810G>A (rs1800280), and c.5234G>A (rs1801187) variants in combination, as shown in Figure 5. For the combination c.2645A>G (rs228406) and c.8810G>A (rs1800280), 66.66% of patients were positive. For the combination c.2645A>G (rs228406) and c.5234G>A (rs1801187), 72.2% of patients were positive. 61.11% of patients were positive for c.8810G>A (rs1800280) and c.5234G>A (rs1801187). c.4375C>T (rs398123953) and c.3406A>T (rs3827462) have not been detected by Sanger sequencing in any of our patients (n = 18 samples).

Combination pattern of variants c.2645A>G (rs228406), c.8810G>A (rs1800280), and c.5234G>A (rs1801187) in 18 patients screened by Sanger sequencing.
Genotype and allele frequency of each variant
The alleles have been reported in reverse orientations. Exon 21 of the DMD gene was screened via Sanger sequencing, and c.2645A>G (rs228406) was detected in all the 18 patients. The homozygous variant G/G was more commonly present in our cohort occurring at frequency of 94.4%. The hemizygous variant G/A was less common, occurring at a frequency of 5.5%. The allele G was present in most patients.
Sanger sequencing of exon 32 of the DMD gene showed an absence of variant c.4375C>T (rs398123953) in 17 patients. The variants could not be detected in one patient due to background noise. The homozygous variant C/C was present in our cohort at frequency of 94.4%. The variant c.4375C>T has been predicted to be pathogenic by the ClinVar database and was present in one patient screened by whole-exome sequencing but was not detected in any patient screened by Sanger sequencing.
The data from Sanger sequencing reported an absence of variant c.3406A>T (rs3827462) in 16 patients in exon 25 of the DMD gene. T > A substitution has not been detected by Sanger sequencing in our cohort. The homozygous variant A/A was present in our patients evaluated by Sanger sequencing at frequency of 88.88%. Due to noise, the variant could not be detected in two patients.
Exon 59 was screened for the variant c.8810G>A (rs1800280) in 18 patients, and 12 patients were found to be positive for it. The expected variant was not detected in five patients by Sanger sequencing. In one patient, the variant could not be detected due to background noise. The homozygous variant A/A was detected in 66.66%, and the homozygous variant G/G was detected in 27.70%. The allele A was present in most patients (66.6%).
Exon 37 was screened for the variant c.5234G>A (rs1801187) in 18 patients, and 13 patients were found to be positive for it. The expected variant was not detected in five patients by Sanger sequencing. The homozygous variant A/A was detected in 66.66%, and the homozygous variant G/G was detected in 27.70%. The hemizygous variant G/A was present in 5.55%. The allele A was present in most patients (69.4%). Figure 6 shows the genotype frequency of each variant, and Table 2 shows the genotype and allele frequency of each variant in our cohort.
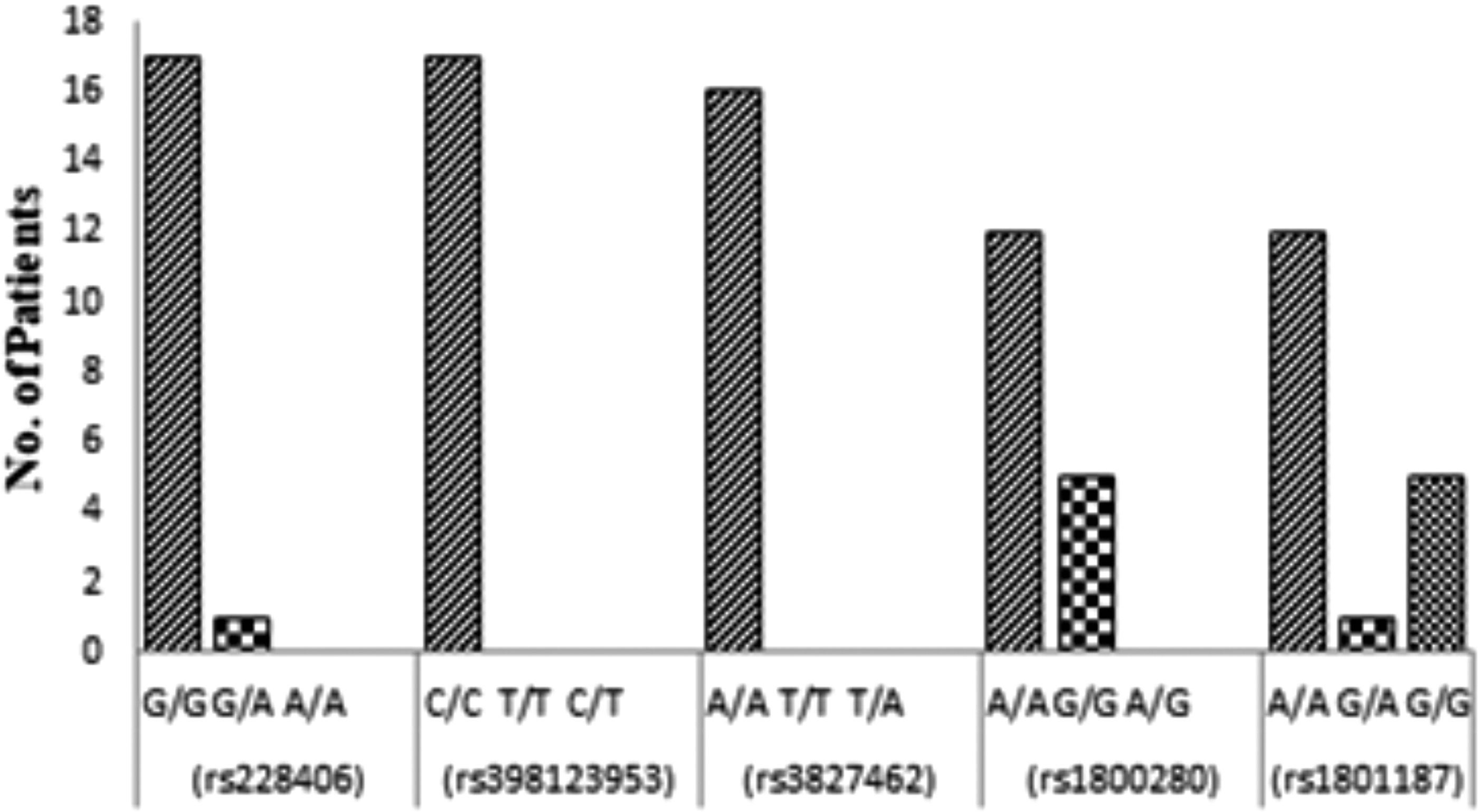
Genotype frequency of each variant screened by Sanger sequencing.
Genotype and Allele Frequency of Each Variant in Our Cohort
Discussion
In this study, we have performed whole-exome sequencing of three MLPA-negative muscular dystrophy patients to determine the spectrum of small mutation in dystrophinopathy patients, identify dystrophinopathy patients who can benefit from Ataluren treatment, determine a definitive genetic diagnosis for muscular dystrophy patients, and identify novel variants. In Pakistan, there are no data available on small mutations in dystrophinopathy patients. Based on the mutations identified by whole-exome sequencing, mutation-specific therapies can be designed for muscular dystrophy patients. Thus, whole-exome sequencing can serve as a diagnostic tool and aid in a precise therapeutic approach.
The study combined whole-exome sequencing with Sanger sequencing to establish a definitive genetic diagnosis for muscular dystrophy patients and to detect the presence of small mutations in the dystrophin gene as identified by whole-exome sequencing in 18 dystrophinopathy patients. The rationale for selecting dystrophinopathy patients for Sanger sequencing is to detect the presence of small mutations in the DMD gene in a Pakistani population, as data on small mutations are typically nonexistent here.
Whole-exome sequencing has not been used for detection of small mutations in Pakistani muscular dystrophy patients. The only work carried out in Pakistan so far is the use of MLPA for detection of large mutations in DMD gene and indirect analysis based on short tandem repeats-based linkage analysis along with serum creatine kinase (SCK) estimation for carrier detection and prenatal diagnosis (Hashim et al., 2011; Ansar et al., 2019). The clinical basis of selecting MLPA-negative muscular dystrophy patients was to exclude dystrophinopathy patients carrying large mutations in the DMD gene, as research on detection of large mutations in the DMD gene has been performed in Pakistan previously (Hassan et al., 2008; Ansar et al., 2019). According to the TREAT-NMD DMD global database, 80% of mutations in the dystrophin gene are large mutations and 20% are small mutations (Bladen et al., 2015). Without the detection of small mutations, appropriate therapeutic strategies cannot be designed. MLPA can detect deletions and duplications in the DMD gene, but it cannot detect small mutations and the location of intronic breakpoints. Utilizing MLPA and Sanger sequencing in combination would involve sequencing of all 79 exons of the dystrophin gene, therefore would be highly laborious and costly. With the use of whole-exome sequencing, small mutations can be detected using a single experimental platform. Therefore, it is recommended to utilize NGS approach for establishing a correct genetic diagnosis of dystrophinopathy patients. Since DMD is a lethal and fatal disease, establishing a correct genetic diagnosis will aid in detecting carrier status of mothers. Prenatal genetic diagnosis and genetic counseling can be performed if a mother is carrier for the disease, helping to reduce the burden of disease in low socioeconomic countries such as Pakistan.
In this study, three MLPA-negative muscular dystrophy patients were screened using whole-exome sequencing, which has identified four missense variants (c.8810 G>A, c.2645A>G, c.5234G>A, and c.3406A>T) and one nonsense variant (c.4375C>T) in the dystrophin gene. This nonsense variant c.4375C>T (rs398123953) in the DMD gene has a phenotypic association with DMD as reported in the Ensemble database and has been classified as a pathogenic variant in the ClinVar database. This mutation is Ataluren specific.
c.572G>T (rs11539445) in exon 1 of the YARS2 gene has been detected in two patients screened by whole-exome sequencing. This variant has also been reported in the LOVD. c.572G>T (rs11539445) variant in YARS2 gene has been associated with MLASA as mentioned in the Ensemble database. Two of our patients had clinical features of muscle weakness particularly involving pelvic girdle with pseudohypertrophy of calf muscles. Based on the genotype-phenotype correlation and after whole-exome sequencing results, two of our patients were found to have MLASA, who were earlier misdiagnosed as cases of DMD based on the initial clinical phenotype.
The five variants in the dystrophin gene detected by whole-exome sequencing have been reported internationally (Luce et al., 2018), but small mutations in the dystrophin gene have not been reported in Pakistani population. We evaluated these variants in 18 dystrophinopathy patients using Sanger sequencing. c.2645A>G (rs228406) was detected in all the 18 patients by Sanger sequencing. It has been reported in the literature as a polymorphism (Luce et al., 2018). c.8810G>A (rs1800280) involving exon 59 of the DMD gene was detected in 12 patients, and c.5234G>A (rs1801187) involving exon 37 of the DMD gene was detected in 13 patients screened by Sanger sequencing. Both these variants have been reported as polymorphisms in an Argentinian dystrophinopathy cohort (Luce et al., 2018). Despite our small cohort, two missense variants were common in two dystrophinopathy patients. Three missense variants have been reported as benign in the ClinVar database. c.5234G>A in exon 37 has been reported as benign in the ClinVar database, but the SIFT information predictor has labeled it to be damaging and PolyPhen has classified it to be probably damaging. All these variants have been reported in the LOVD.
c.4375C>T (rs398123953) and c.3406A>T (rs3827462) were not detected in any of the 18 patients screened by Sanger sequencing. c.3406A>T (rs3827462) has been reported in the LOVD and has been classified as benign by ClinVar.
c.4375C>T (rs398123953) is a nonsense variant that was detected in one patient by whole-exome sequencing. The ClinVar database has reported its association with dilated cardiomyopathy 3B. Nonsense mutation leads to the insertion of premature stop codons. The premature stop codons that might be inserted include TGA, TAG, and TAA. The TREAT-NMD global database detected 4% of mutations with premature stop codon TGA, 3% of mutations with premature stop codon TAG, and 3% of mutations with premature stop codon TAA. All these nonsense mutations would benefit from Ataluren (Bladen et al., 2015). The nonsense mutation could only be detected in one patient in our study, c.4375C>T generated a premature stop codon TGA in our patient.
Conclusions
Our study is the first to detect small mutations in Pakistani muscular dystrophy patients using whole-exome sequencing. We have identified four missense variants (c.8810G>A, c.2645A>G, c.5234G>A, and c.3406A>T) in the dystrophin gene, which have been labeled as polymorphisms in the literature. One nonsense variant (c.4375C>T) in the dystrophin gene was identified, which is amenable to treatment with Ataluren. In addition, c.572G>T (rs11539445) in exon 1 of the YARS2 gene was detected in two patients, and to the best of our knowledge, it is the first time a genetic diagnosis of MLASA patients is established in Pakistani population.
Mutations of YARS2 encoding mitochondrial tyrosyl-tRNA synthetase have been previously reported in patients with infantile- to childhood-onset autosomal recessive MLASA syndrome having myopathic pattern. However, none of our patients showed sideroblastic anemia as has been shown previously owing to molecular heterogeneity (Sommerville et al., 2017; Riley et al., 2018).
Footnotes
Acknowledgment
We acknowledge the Department of Neurology, Dow University of Health Sciences facilitation in patient selection.
Authors' Contributions
M.W. designed and supervised the study. M.Z. collected the clinical data and performed all laboratory procedures. T.F. supervised the PCR experiment. J.A. is the co-supervisor for the study.
Author Disclosure Statement
No competing financial interest exists.
Funding Information
No funding was received for this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.