
Abstract
Background:
Dynein, axonemal, heavy chain 1 (DNAH1) gene mutations have been found to be related to primary ciliary dyskinesia (PCD) and the DNAH1 gene is associated with abnormal flagellar morphology in spermatozoa. Infertility is a common condition in women presenting with primary ovarian insufficiency (POI) characterized by hypergonadotropic hypogonadism. The purpose of this study was to explore the clinical significance of genetic diagnostics in several Chinese primary infertile women with atypical POI.
Methods:
Four atypical POI patients and 100 healthy subjects were recruited, genetic pathogenicityc factors were investigated by whole exome sequencing (WES).
Results:
WES revealed a homozygous deletion mutation in the DNAH1 gene (NM_015512.5; c.11726_11727delCT, p.Pro3909Argfs*33) in one of the four POI patients. The 31-year-old affected woman presented with a normal menstrual cycle and elevated plasma levels of FSH, around the postmenopausal range, but had a normal antral follicle count and normal anti-Müllerian hormone levels. The patient, after two failed ovulation cycles, became pregnant in the third IVF cycle and delivered a healthy girl at term.
Conclusions:
The homozygous deletion mutation in the DNAH1 gene suggested that the patient might have a cilia movement disorder of the fallopian tubes, which is a known infertility factor. Moreover, the significantly elevated plasma level of FSH in this patient is likely one of the most important factors leading to her decreased fertility.
Introduction
Premature ovarian failure (POF) is a complex disease with a complicated clinical presentation. A more scientific and accurate term for the disease is primary ovarian insufficiency (POI), which can be modified appropriately to describe the state of ovarian function (Ferreri, et al., 2020). In recent years, the known etiologies of POI have expanded, although the specific cause of POI in most clinical cases remains unclear (Nelson, 2009).
POI is defined by cessation of ovarian function after puberty but before age 40 years. Typical symptoms of POI are amenorrhea, estrogen deficiency, and substantially elevated levels of serum gonadotrophins (oligo/amenorrhea and increased follicle-stimulating hormone [FSH] concentration [>25 IU/L], recorded at least twice, 4 weeks apart). It has been reported that nearly 1% of women in Western countries with a normal menstrual cycle suffer from POI before reaching age 40 (Coulam et al., 1986; Cooper et al., 2011). Resistant ovarian syndrome (ROS) or “insensitive” ovary syndrome, which is a well-known cause of hypergonadotropic amenorrhea, is related to POF. POI patients have premature depletion of follicles, whereas women with ROS have immature follicles. ROS was first reported in 1969, and is a rare endocrine disorder characterized by hypergonadotropic hypogonadism, and amenorrhea is the most common clinical manifestation (Jones and De Moraes-Ruehsen, 1969). It is well known that many primordial ovarian follicles in ROS are present at ovarian biopsy, consistent with the loss of ovarian sensitivity to gonadotropins.
To understand the possible genetic causes of the condition of the POI patients, mutation screening by whole exome sequencing (WES) was performed. Among the four recruited patients, a homozygous deletion mutation in the dynein axonemal heavy chain 1 (DNAH1) gene, which is related to primary ciliary dyskinesia (PCD) disease, was found in a woman's peripheral blood. The woman suffering from hypergonadotropic hypogonadism who, assisted by in vitro fertilization (IVF), had a successful pregnancy with spontaneous vaginal delivery of a healthy infant at term.
PCD is a genetically heterogeneous autosomal recessive disorder characterized by motile cilia dysfunction. Defective ciliary or flagellar structure and function leads to PCD, which has several clinical manifestations, including chronic otitis media, rhinosinusitis, bronchitis, pneumonia, and male infertility (Zariwala et al., 2007; Leigh et al., 2009; Shapiro et al., 2018). PCD patients exhibit sperm flagellar dyskinesia, and DNAH1 variants leading to PCD were identified among infertile patients with severe asthenozoospermia (Ben Khelifa et al., 2014). However, the relationship between DNAH1 mutation and female infertility has seldom been reported.
Materials and Methods
Human subjects
Four patients with primary infertility and diagnosed with POI were recruited from the Department of Reproductive Medicine, Linyi People's Hospital. All of the patients had a normal karyotype. Peripheral blood samples were taken for DNA extraction from all the patients and all of their family members. One hundred unrelated, anonymous, and fertile women were used as controls.
Our study was conducted at Linyi People's Hospital, China. The ethics committee of Linyi People's Hospital has approved the investigation protocols and written informed consents were obtained from the participants.
Whole exome sequencing
Trio-WES of the patient and her parents was performed. Genomic DNA extracted from peripheral blood for each sample was fragmented to an average size of 180-280 bp and used to create a DNA library following established Illumina paired-end protocols. The Agilent SureSelect Human All ExonV6 Kit (Agilent Technologies, Santa Clara, CA) was used for exome capture according to the manufacturer's instructions. The Illumina Novaseq 6000 platform (Illumina, Inc., San Diego, CA) was used for genomic DNA sequencing to generate 150 bp paired-end reads with a minimum coverage of 97% of the target sequence. After sequencing, base-call file conversion and demultiplexing were performed with the bcl2fastq software (Illumina). The resulting fastq data were analyzed by quality control software to remove low quality reads, and were then aligned to the reference human genome using the Burrows-Wheeler Aligner, and duplicate reads were marked using Sambamba tools. Single nucleotide variants (SNVs) and indels were called with GATK to generate a gVCF file. The raw calls of SNVs and InDels were further filtered with associated thresholds.
Bioinformatic analysis
Filtering of rare variants was performed as follows: (1) variants with an MAF <0.01. (2) Only SNVs occurring in exons or splice sites were further analyzed. (3) Synonymous SNVs that were not relevant to the amino acid alternations were discarded. (4) Variations were screened according to their scores using SIFT, Polyphen, MutationTaster, and CADD software programs. Potentially deleterious variations were retained if the score from more than half of these four software programs supported their potential harmfulness.
To better predict the harmfulness of variation, the classification system of the American College of Medical Genetics and Genomics (ACMG) was used. The variations are classified into pathogenic, likely pathogenic, uncertain significance, likely benign and benign.
Sanger sequencing
Sanger sequencing was performed by polymerase chain reaction (PCR) and direct sequencing of the PCR product on 3500XL Genetic Analyzer (Applied Biosystems, Foster City). Sequences of the primers used for PCR were designed by Primer Premier 5.0 (forward, 5′-CATCTGCATCAGCCAGCTCAAG-3′, reverse, 5′-GTCTTTGTGGGATGGGATGGATTG-3′).
Results
Identification of DNAH1 mutation
We identified a deletion mutation in exon 73 of DNAH1 (NM_015512.5; c.11726_11727delCT, p.Pro3909Argfs*33) was present in an affected female individual (the proband) by using WES. According to the ACMG standards and guidelines, the deletion mutation of DNAH1 (c.11726_11727delCT, p.Pro3909Argfs*33) was “likely pathogenic.”
The pedigree of the family is shown in Figure 1A. Sanger sequencing revealed the heterozygosity of the proband's parents (I-1 and I-2), whereas one unaffected family member (II-2) and healthy controls lacked the variant (Fig. 1B). Thus, the DNAH1 variant showed the predicted segregation pattern for an etiologic variant. Alignment of DNAH1 amino acid sequences in different species, including chimpanzees, mice, and rats, with the use of ClustalW2 showed a premature translational termination codon 33 aa downstream of the mutation at position 3909 (Fig. 1C).
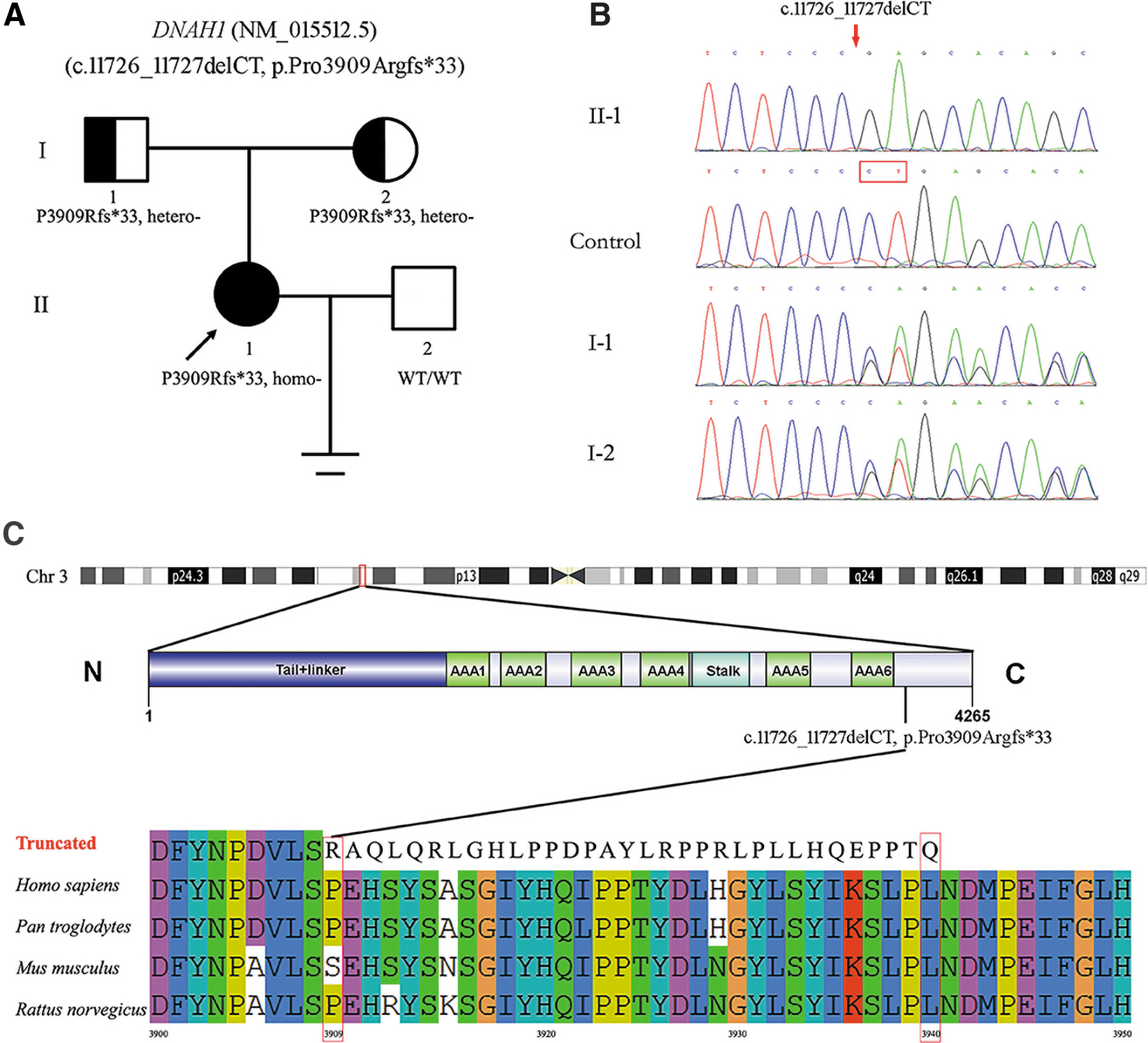
Clinical features of the patient suffered from DNAH1 deletion mutation
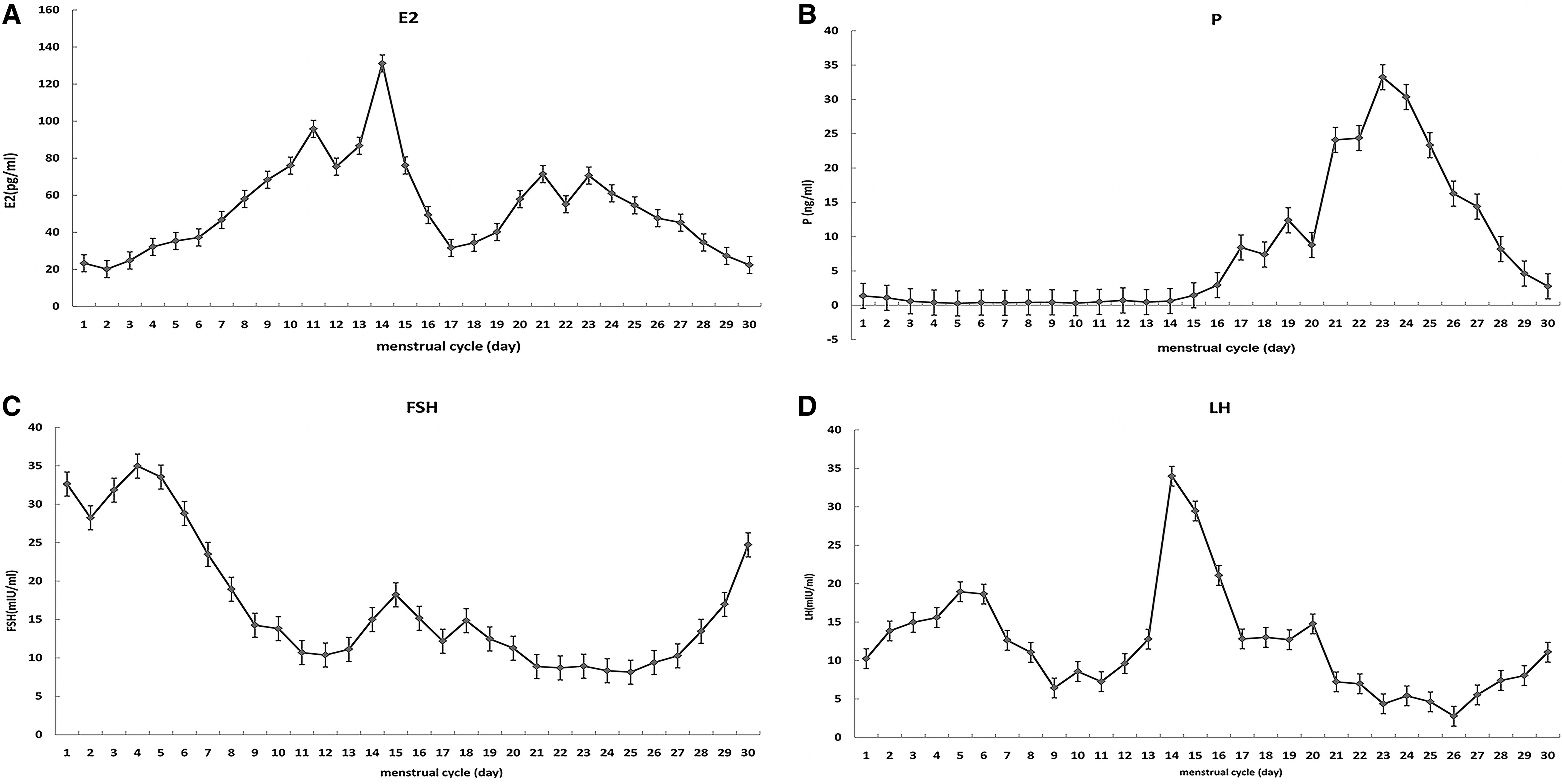
The 31-year-old affected woman with a diagnosis of primary infertility for 3 years was first seen at our institution in April 2011. Salpingography was used to confirm that the patient's fallopian tubes were unobstructed. The male partner showed good sperm quality according to the World Health Organization (2010) criteria. She underwent laparoscopic removal of a chocolate cyst on the right ovary in March 2013. On baseline examination, she had normal ovaries with six to seven antral follicles in the left ovary and seven to eight antral follicles in the right ovary. Before IVF, we measured the hormone levels in the patient's natural menstrual cycle. The plasma hormones were found to be abnormal (Fig. 2).
The patient had undergone three IVF cycles. In the first cycle in April 2014, on day 24 of the cycle when the predominant follicle reached 16.5 mm in diameter, it became a luteinizing follicle. We had to terminate the ovulation cycle.
In June 2014, IVF was planned to be restarted in the luteal phase of the menstrual cycle with an injection of Triptorelin 0.1 mg (triptorelin; Ferring GmbH, Kiel, Germany) subcutaneously once daily for 14 days. When downregulation was achieved on day 5 of the next menstrual cycle, transvaginal sonography (TVS) revealed several growing follicles in both ovaries (in the left ovary eight follicles 5-6 mm in mean diameter, and in the right ovary three follicles with a diameter of 4-7 mm). The E2, P, FSH, and LH serum levels were 39.35 pg/mL, 0.25 ng/mL, 16.4 mIU/mL, and 20.01 mIU/mL, respectively. Human menopausal gonadotropin (Lebaode, Livzon Group, Zhuhai, China) was administrated with an initial dose of 75 U once daily lasted for 10 days and then increased to 225 U once daily for 9 days. Unfortunately, there was no significant change in the growth of antral follicles, and again, the second cycle was also canceled.
The third IVF cycle began in February 2016. On day 15 of the cycle, TVS showed a predominant follicle in the left ovary (17.5 mm), and in the right ovary two follicles of 13.5 and 15.0 mm. However, the E2 level was 129.55 pg/mL, which was lower than on the previous day (139.96 pg/mL). Thirty-six hours after HCG injection, oocyte aspiration was performed. Only one MII egg was obtained and the embryo 4CIII transfer was performed on day 2 following oocyte retrieval. The cycle received luteal phase support with progesterone in the form of vaginal suppositories at a dose of 90 mg once daily (Crinone; Fleet Laboratories Ltd., Northwood, UK). Clinical pregnancy was defined as visualization of a gestational sac and fetal cardiac activity on transvaginal ultrasound following 4-5 weeks of in vitro fertilization and embryo transfer (IVF-ET). Thirty-eight weeks later, a healthy girl baby was born with spontaneous vaginal delivery at term.
Discussion
Causes of POI may be genetic, autoimmune, enzymatic, or iatrogenic (such as from the use of chemotherapeutic drugs; alkylating agent cyclophosphamide, etc.). Women with POI may suffer from night sweats, hot flashes, insomnia, depression, vaginal dryness, dyspareunia, and so on. Timely diagnosis and treatment can reduce the incidence of POI complications in the future (Brand et al., 2013; Faubion et al., 2015; Sullivan et al., 2016). Women with POI should be informed that there is a small chance of spontaneous pregnancy, and there are no interventions that have been reliably shown to increase ovarian activity and natural conception rates (European Society for Human Reproduction and Embryology [ESHRE] Guideline Group on POI et al., 2016). Ovarian failure results in high gonadotrophin levels with little response to human gonadotrophins (Check et al., 2004).
In this study, we illustrate a special case of a woman with high basal FSH concentration. Her AMH was normal, and there were no abnormalities in the number of antral follicles. However, during her natural menstrual cycle, we found that the luteinizing cyst of the dominant follicle was usually accompanied by an increase in the P level. Even after ovulation, one to two luteinizing cysts could be seen in some natural cycles.
In this case, the formation of luteinizing cysts maybe because her basal LH level was too high to produce an effective preovulation endogenous LH surge in the body, resulting in ovulation failure. It suggests that pregnancy may occur in POI women when high FSH concentrations are inhibited by gonadotropin-releasing hormone analogues and low doses of gonadotropin are used to induce ovulation (Check, 2007). However, there are no published prospective trials of sufficient power to support the hypothesis that the reduction in FSH concentrations is more likely to allow the development of follicles than a chance intermittent change in ovarian function.
The current consensus is that POI is characterized by the triad of amenorrhea for at least 4 months in a woman <40 years (Coulam et al., 1986). Strictly speaking, the patient in this study did not conform to the POI standard. She did not have secondary amenorrhea. Instead, her menstrual cycle had been very regular. Perhaps she had a sex hormone-related enzyme abnormality, and we tried to persuade her to undergo a high-resolution array comparative genomic hybridization test to identify pathogenic copy number variations. It has been reported that a deficiency in the DNA damage response may result in chromosomal instability during meiotic and mitotic divisions in the early stages of embryonic development. In addition, pathogenic variants in genes implicated in DNA repair may be associated with ovarian failure (Wood-Trageser et al., 2014).
For the reasons mentioned earlier, trio-WES was conducted to investigate the genetic factors of the patients in our cohort. WES and bioinformatic analysis were performed according to the established protocols in Methods. Given to the characteristics of the pedigree, homozygous and de novo variations were considered to be candidate causal variations in this study. Acquired variants of the proband filtered by a homozygous or de novo pattern were supplied (Supplementary Tables S1 and S2). According to the association of phenotype and genotype, we identified four potentially pathogenic mutations of candidate genes in the proband, including MSH2, RSPH14, CCDC171, and DNAH1. Although the variant of MSH2, p.L481I is not present in gnomAD, mutations in the MSH2 gene mainly result in hereditary nonpolyposis colorectal cancer-1 (HNPCC1). Among RSPH14, CCDC171, and DNAH1, DNAH1 was a relatively defined PCD-associated gene that has been extensively investigated. Moreover, the results showed that no pathogenic mutations associated with POI and hormone production were identified in the patients.
PCD is a clinically and genetically heterogeneous disorder of motile cilia dysfunction with autosomal recessive inheritance, which usually is characterized by various clinical manifestations, including chronic sinusitis, otitis media, and chronic bronchitis (Leigh et al., 2009; Shapiro et al., 2018). It has been reported that DNAH1 variants may leading to immotile sperm flagella which is the cause of male infertility in unrelated males without any other PCD symptoms. (Ben Khelifa et al., 2014; Wang et al., 2017). Further clinical research has shown that PCD-affected female patients can present with subfertility due to defective oviduct cilia (Zariwala et al., 2011).
In this study, the proband had no common PCD manifestations and no pathogenic mutations of PCD-associated genes besides DNAH1. In gnomAD, this DNAH1 frameshift mutation is present in 1:720 individuals in the East Asian group. Moreover, the DNAH1 variant showed the predicted segregation pattern for an etiologic variant (Fig. 1). Thus, DNAH1 mutation may have led to abnormal tubal ciliary movement. The patient underwent laparoscopic removal of a chocolate cyst on the right ovary in 2013. Although no abnormality was found in the fallopian tube during the operation, abnormal tubal ciliary movement was not excluded as an etiology of her infertility. Of interest, Imtiaz et al. presented the only published case where homozygous variant in DNAH1 (c.3460 A ≥ C, p.Lys1154Gln) was judged the most likely genetic cause of female infertility (Imtiaz et al., 2015). In this study, two infertile sisters from a consanguineous Saudi Arabian family were clinically diagnosed PCD and had more severe phenotypes than our case. Owing to PCD is a clinically and genetically heterogeneous disorder, we hold opinion that different variants may result in different degrees of clinical phenotype. In addition, two patients in the study of Imtiaz et al. also carry a potential pathogenic variant (c.668T ≥ C, p.Leu223Pro) of ZMYND10 gene found to be involved in the ciliogenesis and responsible for the cytoplasmic preassembly of ciliary structure elements (Moore et al., 2013; Zariwala et al., 2013).
It is clear from the level hormones in the menstrual cycle and the formation of lutein cysts before ovulation of the patient that the elevated plasma levels of LH have an important impact on the development of follicles and ovulation, which seems to be an important factor in her infertility. Although the patient had elevated FSH levels, few eggs available, and poor embryo quality (4CIII), she achieved successful pregnancy and delivery. It was unknown whether she had suffered from an atypical PCD caused by her DNAH1 mutation, which may lead to a ciliary movement disorder in the fallopian tube cavity of the patient and affect the movement of the fertilized egg in the fallopian tube, leading to implantation failure. When meeting with POI patients in the future, we should consider not only the infertility caused by POI itself, but also other possible factors, such as tubal ciliary dyskinesia.
In conclusion, this article reports a case of successful pregnancy and childbirth with unexplained primary infertility. It has been reported that the elevated FSH level or high early follicular phase serum E2 level predicts a poor pregnancy rate, even in women with regular menses, and when we try to treat women with controlled ovarian hyperstimulation, we may not realize that the induced rise in FSH may downregulate FSH receptors and elicit a poor response (Scott et al., 1990; Toner et al., 1991). In cases such as that presented herein, the natural cycle maybe a good method to obtain an egg. The homozygous deletion mutation in the DNAH1 gene suggested that the patient might have a cilia movement disorder of the fallopian tubes. Further studies are needed to accumulate additional data on mechanisms and treatment modalities for these patients.
Footnotes
Authors' Contributions
M.L. and A.W. designed research. M. L., S.H., X.Z., F.W., D.Z., and X.Z. performed main experiments. X.Z. did the bioinformatic analysis. A.W. and M.L. wrote the article. All authors critically analyzed, discussed, and edited the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The study was supported by the Natural Science Foundation of Shandong Province (ZR2020QH047), the Shandong Provincial Medical Health Science and Technology Development Project (2019WS127), and the National Natural Science Foundation of China (81501620).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.