
Abstract
Purpose:
Familial exudative vitreoretinopathy (FEVR) is a blinding retinal vascular disease. Clinically, FEVR is characterized by incomplete vascularization of the peripheral retina and pathological neovascularization. Only about 50% of FEVR cases can be explained by known FEVR disease gene variations. This study aimed to identify novel genes associated with the FEVR phenotype and explore their pathogenic mechanisms.
Materials and Methods:
Exome sequencing analyses were conducted on one Chinese family with FEVR whose affected members did not exhibit pathogenic variants in the known FEVR genes (verified using Sanger sequencing analysis). Functions of the affected proteins were evaluated using reporter assays. Western blot analysis was used to detect mutant protein expression and the genes' pathogenic mechanisms.
Results:
A rare novel heterozygous variant in DLG1 (c.1792A>G; p.S598G) was identified. The amino acid residues surrounding the identified variant are highly conserved among vertebrates. A luciferase reporter assay revealed that the mutant DLG1 protein DLG1-S598G lost its ability to activate Wnt signaling. Moreover, a knockdown (KD) of DLG1 in human primary retinal endothelial cells impaired tube formation. Mechanistically, DLG1 KD led to a reduction in phosphorylated VEGFR2, an essential receptor for the angiogenic potency that signals the vascular endothelial growth factor molecule.
Conclusions:
The data reported here demonstrate that DLG1 is a novel candidate gene for FEVR.
Introduction
Familial exudative vitreoretinopathy (FEVR, OMIM 133780) refers to a group of inherited retinal diseases that result in severe anomalous retinal vascular development. Criswick and Schepens first described it in 1969 (Criswick and Schepens, 1969). Its clinical appearance varies considerably, and different patients often exhibit different phenotypes, even those from the same family (Gilmour, 2015). The visual defects of FEVR are characterized by vitreoretinal traction, the development of hyperpermeable blood vessels, neovascularization, retinal detachments, and retinal folds (Laqua, 1980). The inherited forms of FEVR can be autosomal dominant (AD), autosomal recessive (AR), or X-linked recessive (XR) (Gow and Oliver, 1971; Laqua, 1980; Plager et al., 1992; Shastry and Trese 1997; Shastry et al., 1997; Crecchio et al., 1998). Mutations in the following have been identified in patients with FEVR: Norrie disease protein (NDP) (Chen et al., 1993), low-density lipoprotein receptor-related protein 5 (LRP5) (Gong et al., 2001; Jiao et al., 2004), frizzled class receptor 4 (FZD4) (Robitaille et al., 2002), tetraspanin 12 (TSPAN12) (Junge et al., 2009; Nikopoulos et al., 2010), zinc finger protein 408 (ZNF408) (Collin et al., 2013), catenin beta 1 (CTNNB1) (Panagiotou et al., 2017), kinesin family member 11 (KIF11) (Robitaille et al., 2014), atonal homolog 7 (ATOH7) (Khan et al., 2012), RCC1 and BTB domain-containing protein 1 (RCBTB1) (Jeng-Hung et al., 2016), exudative vitreoretinopathy 3 (EVR3) on chromosome 11p12-13 (Downey et al., 2001), integrin-18 linked kinase (ILK) (Park et al., 2019), and jagged canonical Notch ligand 1 (JAG1) (Zhang et al., 2020). However, only ∼40-50% of FEVR cases can be explained by these FEVR mutations (Salvo et al., 1937; Kashani et al., 2014; Seo et al., 2015).
The present study identified a novel AD heterozygous missense mutation in DLG1 (c.1792A>G; p.S598G) in a Chinese family affected by FEVR using whole-exome sequencing (WES) analysis. Discs large MAGUK scaffold protein 1 (Dlg1; also called Dlgh1 or SAP97) is an intracellular scaffolding protein with a PEST domain, an L27 domain, three PDZ domains, a SH3 domain, and a GUK domain (Lue et al., 1994; Hering and Sheng, 2002; Godreau et al., 2003; Funke et al., 2005). Endothelial DLG1 plays a role in retinal angiogenesis and BBB/BRB development; it also stimulates β-catenin signaling in CNS ECs (Cho et al., 2019). This study proposed that DLG1 is associated with FEVR—specifically, that DLG1 deficiency might affect angiogenesis by disturbing vascular endothelial growth factor (VEGF) induction.
Materials and Methods
Patient recruitment
This study adhered to the tenets of the Declaration of Helsinki and was approved by the Institutional Ethics Committees of Sichuan Provincial People's Hospital and Xinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine. All the subjects participating in this study signed written informed consent forms. All the experiments were carried out in accordance with the approved study protocols. One Chinese family affected with FEVR was recruited from Xinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine.
Clinical diagnosis
Comprehensive ophthalmic examinations were performed on the proband and proband's parents in the present study. The examinations included optical coherence tomography, B-scan ultrasound, and fundus fluorescein angiography. All the subjects participating in this study met the following inclusion criteria: no premature birth, no drug abuse, and no history of oxygen inhalation.
Mutation screening using WES
Genomic DNA was isolated from peripheral blood and then collected in an EDTA anticoagulant tube. DNA concentration was then measured with an ultraviolet spectrophotometer (NanoDrop ND-2000C; Thermo Fisher Scientific, Allentown, PA). The participants' DNA samples were subjected to WES analysis as described previously (Zhang et al., 2020). The captured library was sequenced on Illumina HiSeq 2500 (Illumina, San Diego, CA). NextGene V2.3.4 software was used to analyze the output sequencing data. Variants were filtered using a public database that included the dbSNP database, the NHLBI Exome Sequencing Program (ESP), gnomAD browser Beta, 1000 genomes, ExAC browser (Beta), and 1600 in-house non-FEVR controls.
Mutation validation
The mutations revealed from WES were confirmed by Sanger sequencing. PCR primers were designed by Primer3 Input (list included in Supplementary Table S1). According to the manufacturer's instructions, the DNA samples were subjected to Sanger sequencing on an ABI 3730 automatic sequencer (Applied Biosystems, Foster City, CA).
Construction of expression plasmids
N-terminal HA-tagged DLG1 expression vectors were purchased from Youbao Biotechnology, Inc., (Changsha, Hunan, China). A Q5® Site-Directed Mutagenesis Kit (NEB, Ipswich, MA) was used to construct the missense variant into wild-type (WT) DLG1 complementary DNA (cDNA). According to a previous description, FZD4, NORRIN, and TAPAN12 expression plasmids were provided by Dr. Jeremy Nathans (Xu et al., 2004). Before use, these plasmids were verified via Sanger sequencing.
Cell culture
This study used HEK/293T cells (ATCC CRL-3216), HEK293STF (HEK293 cells the Wnt/b-catenin reporter SuperTOPFlash) (Xu et al., 2004), and human primary retinal endothelial cells (HREC) (Cell Systems, Seattle, WA). HEK/293T cells and Super TOP Flash (STF) cells were cultured with DMEM supplemented with 10% fetal bovine serum. HRECs were grown in EGM™-2 media (Lonza, Rochester, NY). All cells were cultured in a 5% CO2 incubator at 37°C.
Luciferase assays
One hundred nanograms of NORRIN, 100 ng of FZD4, 100 ng of TAPAN12, 15 ng DLG1 (wild type or mutants), and 100 ng of Renilla luciferase plasmid (pRL-TK) control vector ((Promega, Madison, WI) were cotransfected into each well of a 24-well plate seeded with HEK293STF. Lipofectamine 3000 Transfection Reagent (Invitrogen, CA) was used to transfect the plasmid into cells. After 48 h, a TransDetect® Double-Luciferase Reporter Assay Kit (TransGen Biotech, Beijing, China) was used to assay the luciferase activity of the transfected cells. Normalizing Firefly/Renilla values were used to calculate relative luciferase activity.
Knockdown of DLG1
The DLG1 shRNA (5′-GCACAGATGCAGATTATGAAT-3′) or the negative control shRNA (5′-TTCTCCGAACGTGTCACGT-3′; Genechem, Shanghai, China) carried by lentivirus were transfected into HRECs at passages 3-7; expression of DLG1 was then evaluated by quantitative reverse transcription PCR (RT-qPCR).
Reverse transcriptional polymerase chain reaction and RT-qPCR
The entire HREC RNA was extracted using RNeasy Mini Kits (QIAGEN, Dusseldorf, Germany). EasyScript One-Step reverse transcription polymerase chain reaction (RT-PCR) SuperMix (TransGen Biotech) was used to reverse-transcribe the RNA. The cDNA was then used as a template for PCR. TransStart Tip Green qPCR SuperMix (TransGen Biotech) was used in a 7500 Real-Time PCR System (Applied Biosystems) to amplify the cDNA. All the primers used in this study are listed in Supplementary Table S1.
Western blotting
HEK/293T cells transfected with DLG1 (vector, wild type, or mutant) and HRECs (control or shDLG1) were lysed in 1 × RIPA buffer containing protease inhibitor (Roche) and sonicated for 5 s; this was repeated three times. The lysed proteins (20 μg) were loaded into a 10% polyacrylamide gel and transferred to NC membranes (EMD Millipore HATF00010, MA) for immunoblotting. All the antibodies used in the study are listed in Supplementary Table S2. A Matrigel tube formation assay was performed as described in Zhang et al. (2019).
Results
Clinical evaluation
The proband developed eye problems in early childhood. A B-scan ultrasound of the proband revealed cloudy vitreous bodies with visible bands in both eyes (Fig. 1A). The proband's mother, also affected with eye problems, showed severe clinical features, including total loss of visual acuity in the left eye (Fig. 1B) and retinal detachment in the right eye (Fig. 1C).
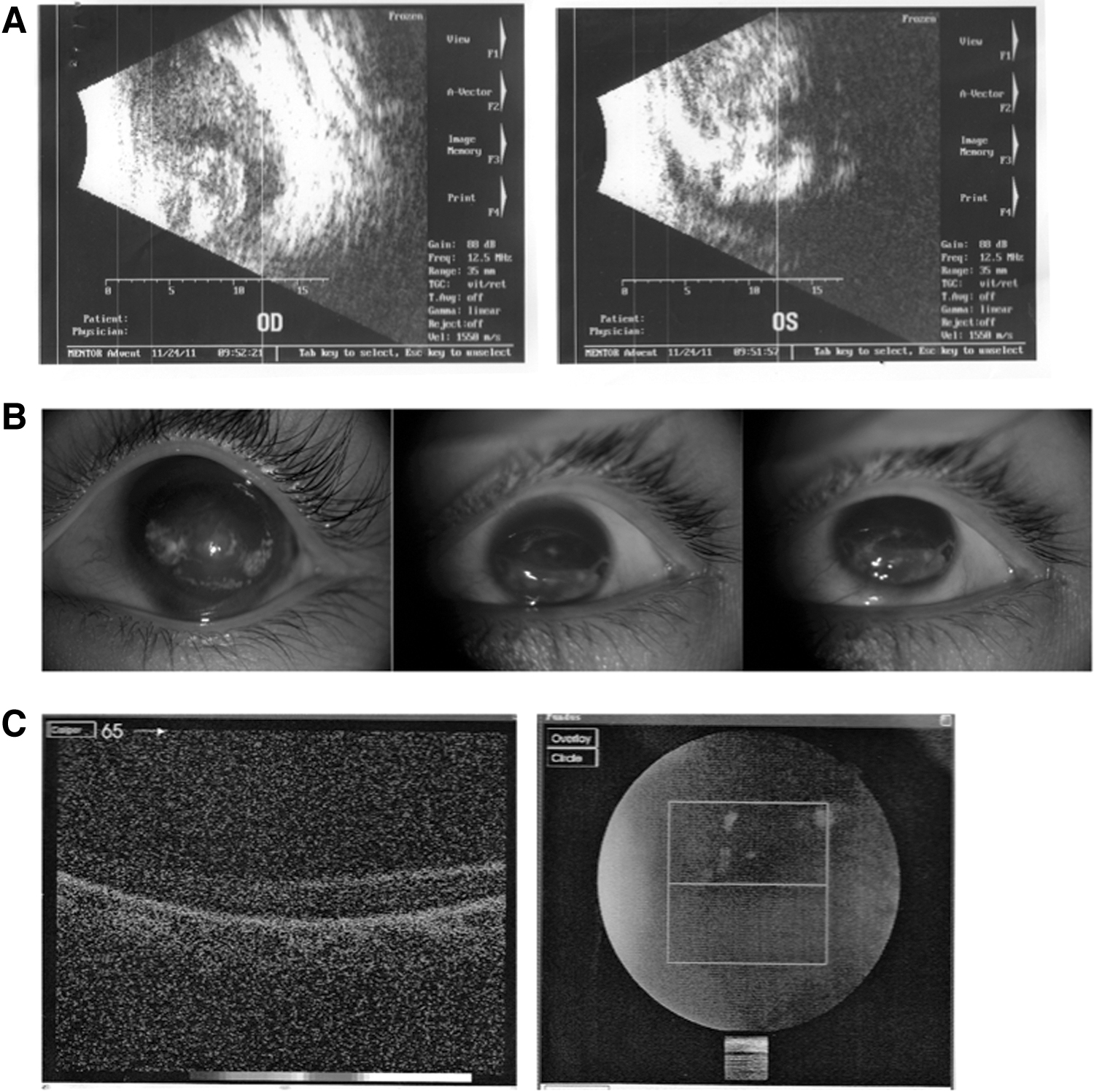
Clinical evaluation of patients with FEVR.
Heterozygosis variant in DLG1 as a candidate mutation
To determine the pathogenic mutation in the family with FEVR, the genomic DNA sample of the proband was subjected to WES analysis. One novel heterozygous variant c.1792A>G (p.S598G) in the DLG1 gene was identified (Fig. 2A, B). Sanger sequencing analysis was performed on DNA samples from the proband and his parents to validate the mutations. The affected mother of the proband also carried the c.1792A>G mutation, whereas the unaffected father did not (Fig. 2A, B). This mutation has not previously been reported in FEVR cases; this study used online tools to predict that the mutation would be damaging (Supplementary Table S3). The frequency of c.1792A>G in the Genome Aggregation Database (gnomAD) was 0.000003982 (1/251164); no homozygous mutation was identified in the database. In addition, the mutation was absent in 1600 ethnic-matched controls. Furthermore, the amino acid residues surrounding the mutation are highly conserved from Homo sapiens to Xenopus tropicalis (Fig. 2C). Taken together, these data indicate that c.1792A>G (p.S598G) in the DLG1 gene is a candidate mutation in the FEVR family.

DLG1 gene variant identified in the FEVR family.
The S598G variant negatively impacts DLG1 function
DLG1 has been reported to play an essential role in vascular development via interaction with the FZD4 receptor of the Wnt signaling (Cho et al., 2019). In the present study, site-directed mutagenesis was used to evaluate the impacts of the variant on the DLG1 protein. When probing with HA antibodies, a unique band at about 120 kDa appeared in the western blotting (WB) results. There was no significant change in the expression levels of the mutant protein compared with the WT protein (Fig. 3A). Top-flash luciferase assay was carried out in the HEK293STF cells to investigate the pathogenicity of the mutations. The results showed that S598G lost its ability to enhance NORRIN/β-catenin signaling (Fig. 3B). These data suggested that the variant (c.1792A>G, p.S598G) negatively affected DLG1 protein activity.
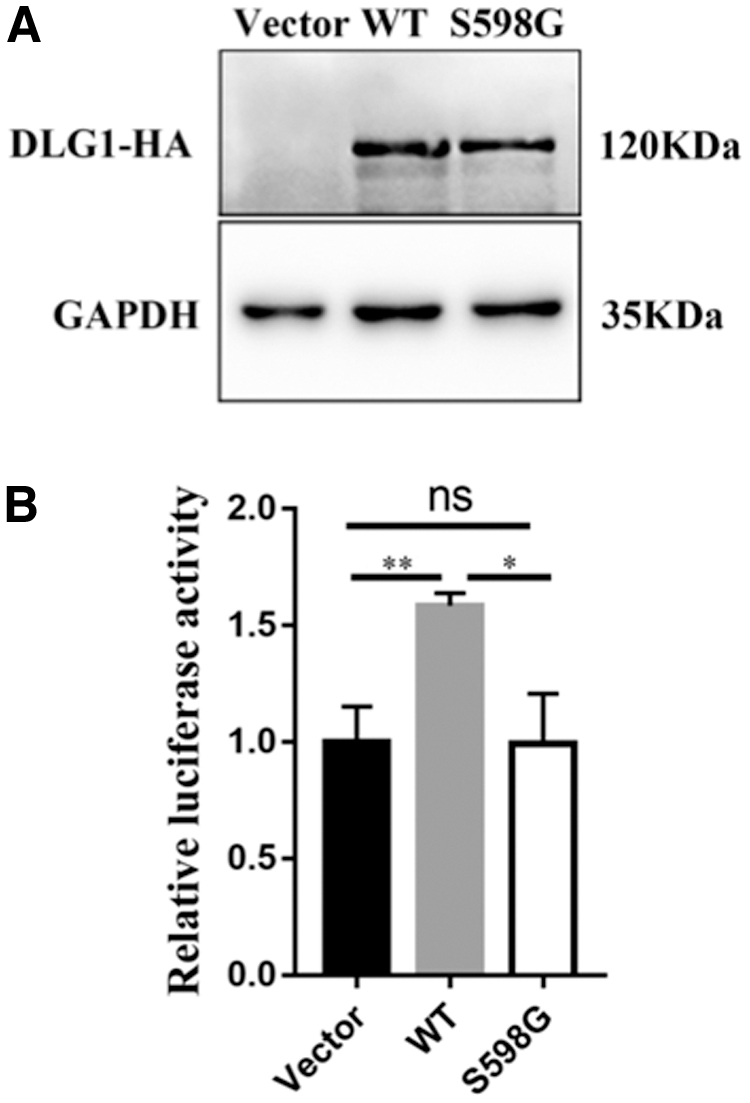
DLG1-S598G mutant protein failed to activate Norrin-β-catenin signaling.
Tube formation impaired in shDLG1 HRECs
The study then investigated whether DLG1 affected endothelial cell function in vitro. RT-PCR analysis revealed that DLG1 was expressed in HRECs (Fig. 4A) and in various tissues of mice (Fig. 4B). A lentivirus-directed shRNA knockdown (KD) system was applied to deplete the expression of DLG1 in HRECs (shDLG1 HRECs). The RT-PCR results revealed that the mRNA level of DLG1 in HRECs was reduced to about 40% compared to the level in the control cells (Fig. 4C). Then, a Matrigel-based in vitro tube formation assay revealed compromised tube formation in shDLG1 HRECs compared with shCtrl HRECs after 3 or 6 h (Fig. 4D, E).
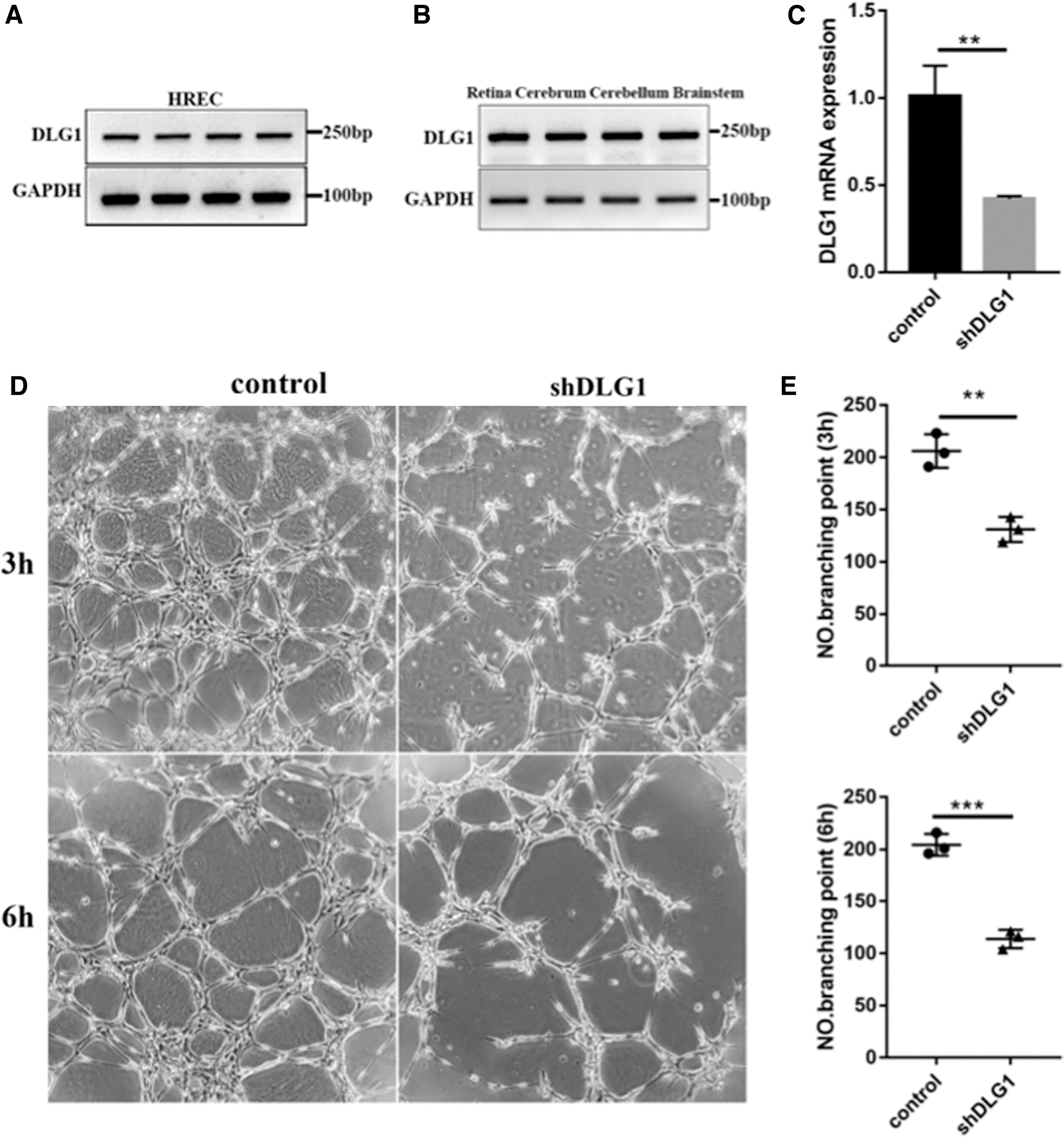
Reduction of DLG1 expression impaired tube formation in HREC.
Reduction of DLG1 impaired VEGFR2 phosphorylation
WB analysis was performed in the HRECs with DLG1 KD to detect the mechanism underlying FEVR caused by the DLG1 mutation. Compared to their WT controls, the phosphorylation (at Tyr1175) level of VEGFR2 was significantly reduced (Fig. 5A, B). An essential component of angiogenesis, the VEGF ligand binds to the VEGFR2 receptor and stimulates the development of endothelial tip cells. Therefore, DLG1 controls angiogenesis via regulating the phosphorylation level of VEGFR2.
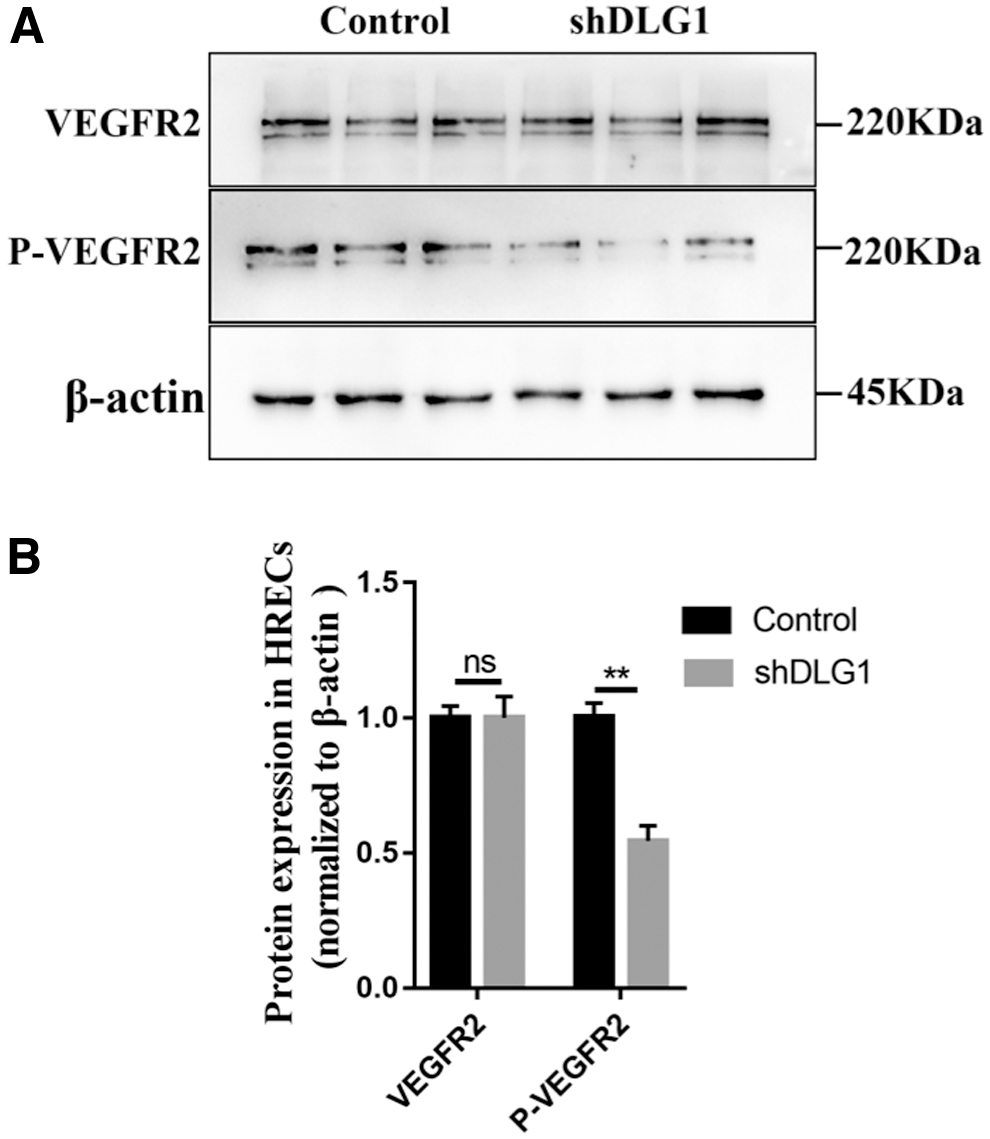
Reduction of DLG1 expression resulted in decreased phosphorylation level of VEGFR2 protein.
Discussion
DLG1 is a molecular scaffolding protein; it is a key regulator of tissue growth, cell architecture, differentiation, and plasticity cell migration (Won et al., 2017). DLG1 also plays crucial roles in adherens junction integrity and differentiation in mammalian epithelial cells (Laprise et al., 2004). Previous studies have indicated that DLG1 regulates retinal angiogenesis and the blood-retina and blood-brain barriers by activating beta-catenin signaling (Cho et al., 2019). This study identified one heterozygous pathological variant, c.1792A>G (p.S598G), in DLG1 in nonsyndromic FEVR patients and demonstrated that the deletion of DLG1 in HERCs led to angiogenesis defects.
The variant c.1792A>G (p.S598G) is located in the Src homology 3 (SH3) domain of DLG1 (Fig. 2D). SH3 is the protein interaction domain that binds to proline-rich ligands (Godreau et al., 2003; Marcello et al., 2007). In some tyrosine kinases, the SH3 domain plays a role in mediating protein-protein interactions and regulating protein functions through intramolecular interactions (Andreotti et al., 1997; Moarefi et al., 1997). The SH3 domain also intramolecularly associates with the GUK domain (Kim et al., 1997; Hanada et al., 2000; McGee et al., 2001). Genetic studies have determined that the SH3 domains of MAGUKs are essential for growth regulation and epithelial structure (Woods et al., 1996; Hough et al., 1997). All the genetic mutations in the SH3 domain previously identified in DLG1 disrupted intramolecular binding (McGee and Bredt, 1999). SH3 mutation resulted in a relocalization of DLG1 to the nucleus (Kohu et al., 2002). The p.S598G mutation identified in this study might impair the interaction of the SH3 domain with other binding partners or the relocalization of DLG1.
VEGF is a key player governing angiogenesis, and VEGFR2 is the main receptor for VEGF-induced signal transduction in endothelial cells. When bound to ligands, VEGFR2 autophosphorylates and is activated (Meyer, 2014). VEGFR2 signal transduction is essential for achieving proliferation, chemotaxis, and sprouting caused by VEGF stimulation as well as the survival of cultured endothelial cells in vitro and angiogenesis in vivo (Karkkainen and Petrova, 2000; Rahimi et al., 2000; Claesson-Welsh, 2003). The Tyr1175 phosphate of VEGFR2 can bind phosphatidylinositol 3-kinase (Holmqvist et al., 2004). Further, DLG1 can cause the carcinogenic activation of PI3K (Frese et al., 2006). In the present study, the phosphorylation (at Tyr1175) level of VEGFR2 was significantly reduced when DLG1 was knocked down. Thus, this study proposes that the loss of DLG1 affects angiogenesis by reducing the Tyr1175 phosphate of VEGFR2.
In conclusion, this study identified one novel heterozygous mutation (c.1792A>G, p.S598G) in DLG1 in a FEVR family; it demonstrates that DLG1 is a novel FEVR candidate gene. Further investigation with more FEVR cases to validate this DLG1 pathogenesis is warranted.
Footnotes
Acknowledgments
The authors want to thank all the patients and their family members for participating in this study.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research project was supported by the National Precision Medicine Project (2016YFC0905200), the National Natural Science Foundation of China (81790643 to Z.Y.), (81470668, 81770950 to XJ.Z.), (82000913 to S.L.), (81770963 to P.F.), (81770964, 81470642 to P.Z.); Grant from Chinese Academy of Medical Sciences (2019-12M-5-032), the Department of Science and Technology of Sichuan Province, China, (2020JDZH0027, 2018JZ0019 to X.J.Z.), (2020ZYD037 to Z.Y.), the non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2019PT3100200).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.