
Abstract
Background:
Periodontitis is a multifactorial disease mainly caused by the formation of plaque biofilm, which can lead to the gradual destruction of tooth-supporting tissues. Current research on the genetics and epigenetics of periodontitis remains relatively limited, and the molecular mechanisms remain largely unknown.
Objective:
Our aims were to construct competitive endogenous RNA (ceRNA) network and determine DNA methylation patterns of target genes to help elucidate the pathogenesis of periodontitis.
Methods:
We analyzed the expression profiles of the GSE16134, GSE54710, GSE10334, and GSE59932 datasets from the Gene Expression Omnibus database through the weighted gene coexpression network analysis system. In addition we screened mRNAs that are regulated by the level of methylation and that are associated with the occurrence of periodontitis. Next, a lncRNA-miRNA-mRNA ceRNA network was constructed using databases including miRanda and TargetScan. Gene ontology and Kyoto Encyclopedia of Genes and Genomes enrichment analyses were conducted for genes in the clinically significant modules. Finally, a protein-protein interaction network was built.
Results:
We identified four mRNAs, four miRNAs, and six lncRNAs as shared differentially expressed genes related to the periodontitis inflammation pathway. IL-6, IFNA17, CXCL12, and TNFRSF13C were identified as key genes whose expression was significantly enriched in the nuclear factor κB and TLR4 pathways. Moreover, the expression of 28 genes were downregulated by hypermethylation and 70 genes were upregulated by hypomethylation.
Conclusions:
The constructed ceRNA network can improve our understanding of the pathogenesis of periodontitis. Candidate mRNAs from the ceRNA network could serve as new therapeutic targets and prognostic biomarkers in periodontitis.
Introduction
Periodontitis (Chronic periodontitis, CP) is the most common chronic infectious disease that occurs in periodontal supporting tissues supporting tissues. Clinically, it mainly presents as gingival inflammation and progressive absorption of the alveolar bone. Owing to the long-term lack of oral health awareness and delay in treatment, it can eventually lead to tooth loss, reduction in mastication, and even endangering the overall health status of the patient. The etiology of periodontal disease involves many factors such as infection, immunity, race, heredity, daily habits, environment, and psychology (Williams et al., 2008). In recent years, more scholars have recognized that genetic and epigenetic factors can affect the progress of periodontitis by regulating the host immune response caused by periodontal pathogens (Zhang et al., 2010a). To clarify the etiological mechanism behind periodontitis, it is particularly important to determine the changes of genetic and epigenetic factors in the pathogenesis of the disease.
Noncoding RNA, as an important regulatory factor in epigenetics, has two main types, including microRNAs (miRNAs) and long noncoding RNAs (lncRNAs). MicroRNA contains 22 base pairs and can negatively regulate the expression of messenger RNA (Saito et al., 2017). Recent studies using chip analysis (Saito et al., 2017) have shown that there is a significant difference in the expression profile of microRNA between inflamed and healthy gingival tissue in patients with periodontitis. Long-chain noncoding RNA, as a type of noncoding RNA, contains >200 base pairs and can regulate the expression of messenger RNA encoding proteins in a positive or negative manner. Studies have shown that long-chain noncoding RNAs are differentially expressed in the inflamed gingival tissues of patients with CP (Zou et al., 2015). DNA methylation is a core epigenetic modification pathway.
Many studies have shown that DNA methylation can cause changes in chromatin structure, DNA conformation, DNA stability, and the interaction between DNA and proteins, thereby controlling gene expression (Michalak et al., 2019). Some studies have shown that, compared with the corresponding genes in healthy individuals, genes encoding proinflammatory cytokines have lower levels of DNA methylation in patients with chronic and aggressive periodontitis, which lead to overexpression in inflammatory tissues (Oliveira et al., 2009; Andia et al., 2010; Zhang et al., 2010b; Ishida et al., 2012).
The results of using RNA chip analysis methods to study the expression of various RNAs in periodontitis have great heterogeneity (Schmalz et al., 2016). Therefore, it is necessary to use bioinformatics technology to integrate all the chip datasets in these previous studies to predict important biological markers related to the risk of periodontitis.
This study uses bioinformatics methods, based on high-throughput RNA sequencing and microarray data of periodontitis, and integrates the expression profile data of long-chain noncoding RNA, microRNA, and messenger RNA to construct a competition endogenous RNA that participates in the pathogenesis of periodontitis. Moreover, combined with the study of DNA methylation, we have a deeper understanding of the molecular mechanisms related to the development of periodontitis.
Materials and Methods
Data downloading and preprocessing
All microarray data were downloaded from the GEO database (www.ncbi.nlm.nih.gov/geo/), representing the largest resource of public microarray data. Screening criteria were as follows: (1) periodontitis; (2) human gingival samples; and (3) number of samples >10. According to the above criteria, GSE16134, GSE54710, GSE10334, and GSE59932 were included in the study.
The gene expression microarray (GSE16134) contained 241 periodontitis samples and 69 normal gingival tissues, based on the GPL15159 platform. The miRNA microarray (GSE54710) included 159 periodontitis samples and 41 normal gingival samples, based on the GPL15159 platform. The lncRNA microarray (GSE10334) included 183 periodontitis samples and 64 normal gingival samples based on the GPL570 platform. The methylation microarray (GSE59932) included 10 periodontitis samples and 12 normal gingival samples, based on the Illumina HumanMethylation450 BeadChip (GPL13534 platform). The details of these microarray data are listed in Table 1. R software (version 3.5.1) was used to perform the bioinformatics analysis. The Limma package in R (Ritchie et al., 2015) was used to perform quantile normalization and difference analysis on the obtained microarray data.
Basic Information on Gene Expression Profiling
Weighted gene coexpression network construction and module identification
We used the weighted gene coexpression network analysis (WGCNA) R package to build a coexpression network. Before the network was constructed, we clustered the samples to evaluate whether there were any obvious outliers. Then, we used a sample dendrogram and trait heat map for visualization. We constructed an adjacency matrix to calculate gene-gene interactions. Calculating the connection coefficient of the gene pair was based on the following formula.

where aij is the adjacency function between the i-th and j-th genes, and Sij is the Person correlation coefficient of genes i and j.
Second, the pickSoft Threshold function of WGCNA was used to calculate the soft thresholding power β. Third, a topological overlap matrix (TOM) similarity function was used to convert the adjacency matrix to a topological matrix. Finally, hierarchical clustering and the dynamic tree cut function were used to generate the modules. The formula for TOM is as follows:

Among them,  represents for any node u that is commonly connected with gene i and gene j. And
represents for any node u that is commonly connected with gene i and gene j. And  , which represents the sum of the adjacency coefficients of all nodes u connected to gene i, which can be regarded as the correlation of the common adjacency coefficients.
, which represents the sum of the adjacency coefficients of all nodes u connected to gene i, which can be regarded as the correlation of the common adjacency coefficients.
Construction of module-trait relationships
Gene modules highly correlated with periodontitis traits, module eigengenes (MEs) were identified. The ME represents the first main component of the module, and the expression of ME represents all genes in each module. Module membership (MM) represents the correlation between gene and the clinical status of periodontitis. Clinically significant modules were identified by calculating the correlation between MEs and periodontitis traits. Gene significance and MM were calculated to relate modules to clinical traits. For further analysis, we extracted the corresponding module gene information and visualized the network of eigengenes.
Identification of hub methylated sites
Methylated mRNA was analyzed according to the results of the coexpression analysis. Negatively and positively correlated mRNAs were selected at hypermethylated and hypomethylated sites, respectively. Subsequently, the online Venn diagram tool was used for visualization.
ceRNA network construction
To shed light on the interactions between mRNA-miRNA and miRNA-lncRNA, we used the following five databases: miRMap (Vejnar et al., 2013), miRanda (John et al., 2004), miRDB (Wong and Wang, 2015), TargetScan (Agarwal et al., 2015), and miTarBase (Hsu et al., 2011). The criterion for predicting the interaction between miRNA and mRNA was that each target mRNA appeared in three databases simultaneously. Two separate ceRNA networks were constructed using the interactions of mRNA-miRNA and miRNA-lncRNA, and the upregulated and downregulated RNAs were visualized using the online bioinformatics database Cytoscape v3.7 (Shannon et al., 2003).
Analysis of function and pathway enrichment
We used the Database for Annotation Visualization and Integrated Discovery (DAVID) to analyze gene ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) to analyze pathway enrichment and explore gene functions and signaling pathways. The GO analysis included molecular function (MF), biological process (BP), and cellular component (CC). We used clusterProfiler in R software and took mRNA in the ceRNA network to perform GO and KEGG enrichment analyses. The results were visualized using the “Dot plot” and “pathview” functions in R.
Protein-protein interaction network construction
To construct the protein-protein interaction (PPI) network, we input the ceRNA network genes into the Search Tool for the Retrieval of Interacting Genes (STRING) database (www.string-db.org), which is a database for predicting protein interactions and can provide important insights into the underlying mechanisms of periodontitis. We selected a required confidence (combined score) >0.4 as the threshold of PPI.
Results
Construction of a weighted coexpression network and identification of key modules correlating with periodontitis
The complete workflow of the research is given in Figure 1. We downloaded the following four gene chip datasets of periodontitis from the GEO database: GSE16134, GSE54710, GSE10334, and GSE59932. After standardizing the raw data of GSE16134, WGCNA was performed on 310 samples. The flashClust function was used to exclude abnormal samples (Supplementary Fig. S1).

Flowchart of the research protocol. Four datasets of the GEO database were used to conduct the mRNA-methylation analysis and a ceRNA network. According to the enrichment analysis results, the subnetwork was constructed. Color images are available online.
Choosing an appropriate soft threshold power is a key step in constructing the WGCNA; when the soft threshold power β was set to 8, the scale-free topology fitting index reached 0.813 (Supplementary Fig. S2). The same method was used to perform WGCNA on the GSE54710, GSE10334, and GSE59932 samples. In total, 16 coexpressed gene modules were identified using the dynamic tree cutting method. Among them, the largest module comprised 1005 genes (midnight blue) and the smallest module comprised 36 genes (dark orange). Genes in gray modules indicate that they are not included in any module (Fig. 2a). In GSE54710, the largest module comprised 433 genes (turquoise) and the smallest module comprised 31 genes (magenta) (Fig. 2b). In GSE10334, the largest module comprised 535 genes (turquoise) and the smallest module comprised 30 genes (green) (Fig. 2c). In GSE59932, the largest module comprised 29004 genes (darkgoldenrod4) and the smallest module comprised 52 genes (goldenrod4) (Fig. 2d).
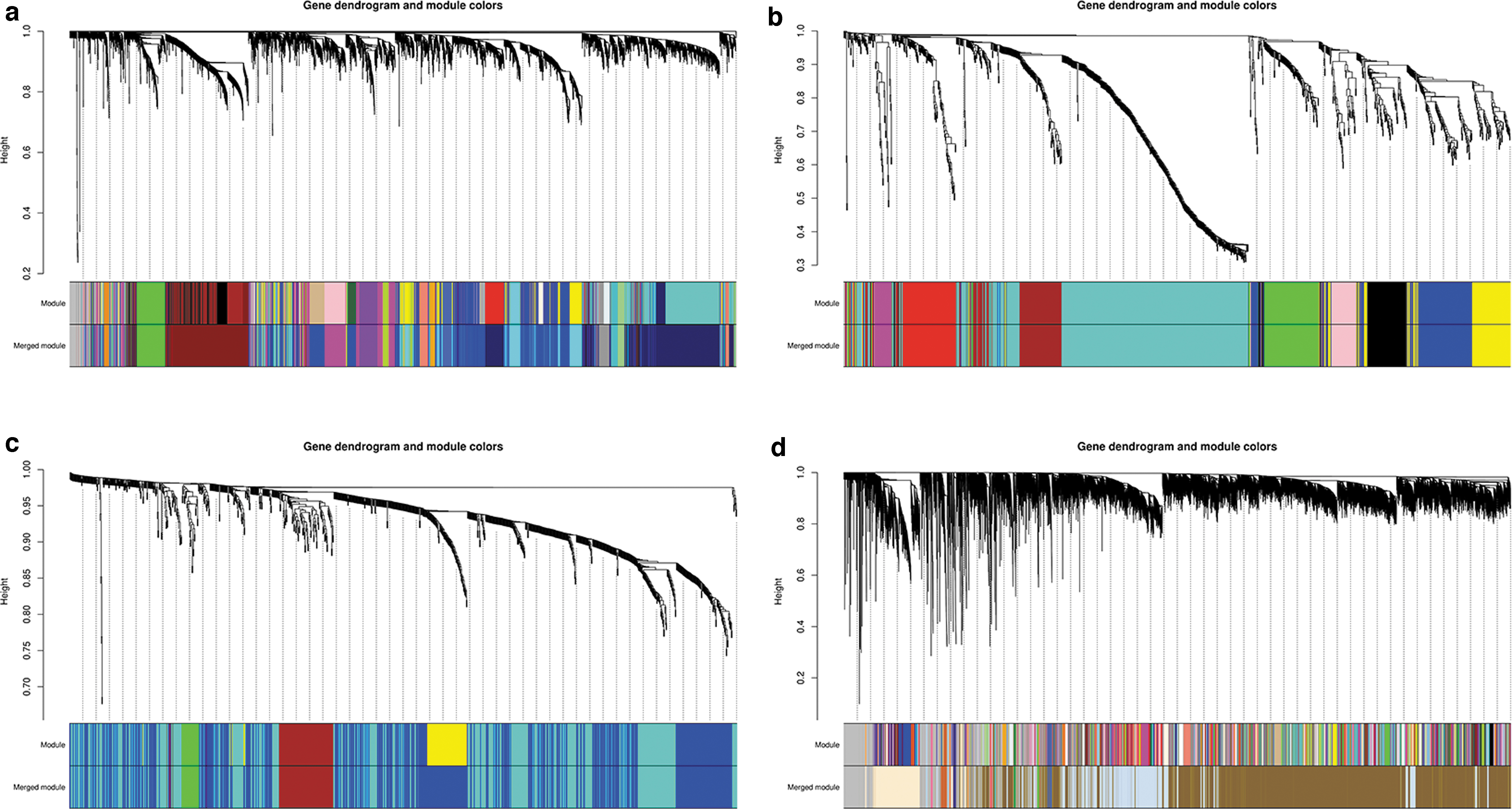
Clustering dendrograms of
We next determined the relationship between the modules by drawing the eigengenes adjacency heatmap, which describes the TOM between all genes included in the analysis; the results indicated that the gene expression was highly independent between modules (Fig. 3a-d).
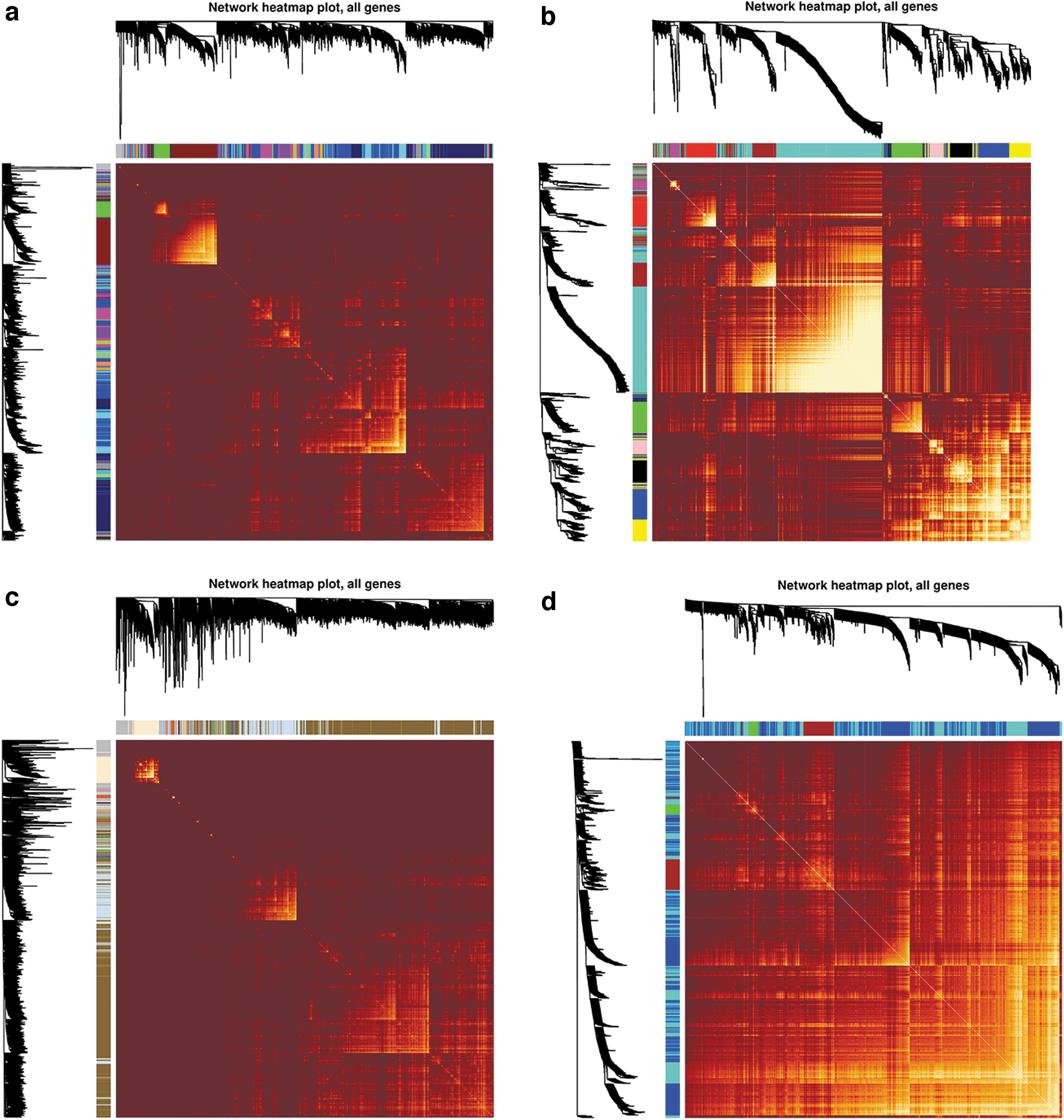
Visualization of the WGCNA network using a heatmap plot. Clustering heatmap of
We analyzed the connectivity of eigengenes. The eigengene is defined as the first principal component of a given module. It can be considered a representative of the gene expression profiles in a module. The results showed that the three combinations (dark turquoise and light green modules, green-yellow and magenta modules, and cyan and royal blue modules) had a high degree of interconnectivity in GSE16134 (Fig. 4a). In GSE54710, three combinations (brown and turquoise modules, green and yellow modules, and black and blue modules) have a high degree of interconnectivity (Fig. 4b). In GSE10334, green and blue modules have moderate interconnectivity (Fig. 4c). In GSE59932, blanchedalmond and burlywood modules have a high degree of interconnectivity (Fig. 4d).

Finally, we used Pearson's correlation analysis to correlate the modules with clinical characteristics. In GSE16134, the results showed that the salmon modules were significantly positively correlated with periodontitis status (R = 0.22, p = 1e-04), and the darkred module was negatively correlated with periodontitis status (R = −0.43, p = 1e-15) (Fig. 4e). In GSE54710, the blue modules were significantly positively correlated with periodontitis status (R = 0.25, p = 3e-04), and the red module was negatively correlated with periodontitis status (R = −0.44, p = 1e-10) (Fig. 4f). In GSE10334, the blue modules were significantly positively correlated with periodontitis status (R = 0.28, p = 8e-06), and the turquoise module was negatively correlated with periodontitis status (R = −0.5, p = 7e-17) (Fig. 4g). In GSE59932, the lightsteelblue1 modules were significantly positively correlated with periodontitis status (R = 0.83, p = 2e-06), and the darkgoldenrod4 module was negatively correlated with periodontitis status (R = −0.79, p = 1e-05) (Fig. 4h).
Relationship between differentially methylated genes and differentially expressed genes
According to the results of the WGCNA using the GSE16134 dataset, we identified 118 upregulated differentially expressed genes (DEGs) and 532 downregulated DEGs in the salmon module and the dark red module, respectively. In GSE59932, there were 5718 hyper-differentially methylated genes (DMGs) and 20,041 hypo-DMGs in the light steel blue module and the dark goldenrod4 module, respectively.
Subsequently, we selected negatively and positively correlated mRNAs at hypermethylated and hypomethylated sites, respectively, and constructed the Venn diagram (Fig. 5a, b).
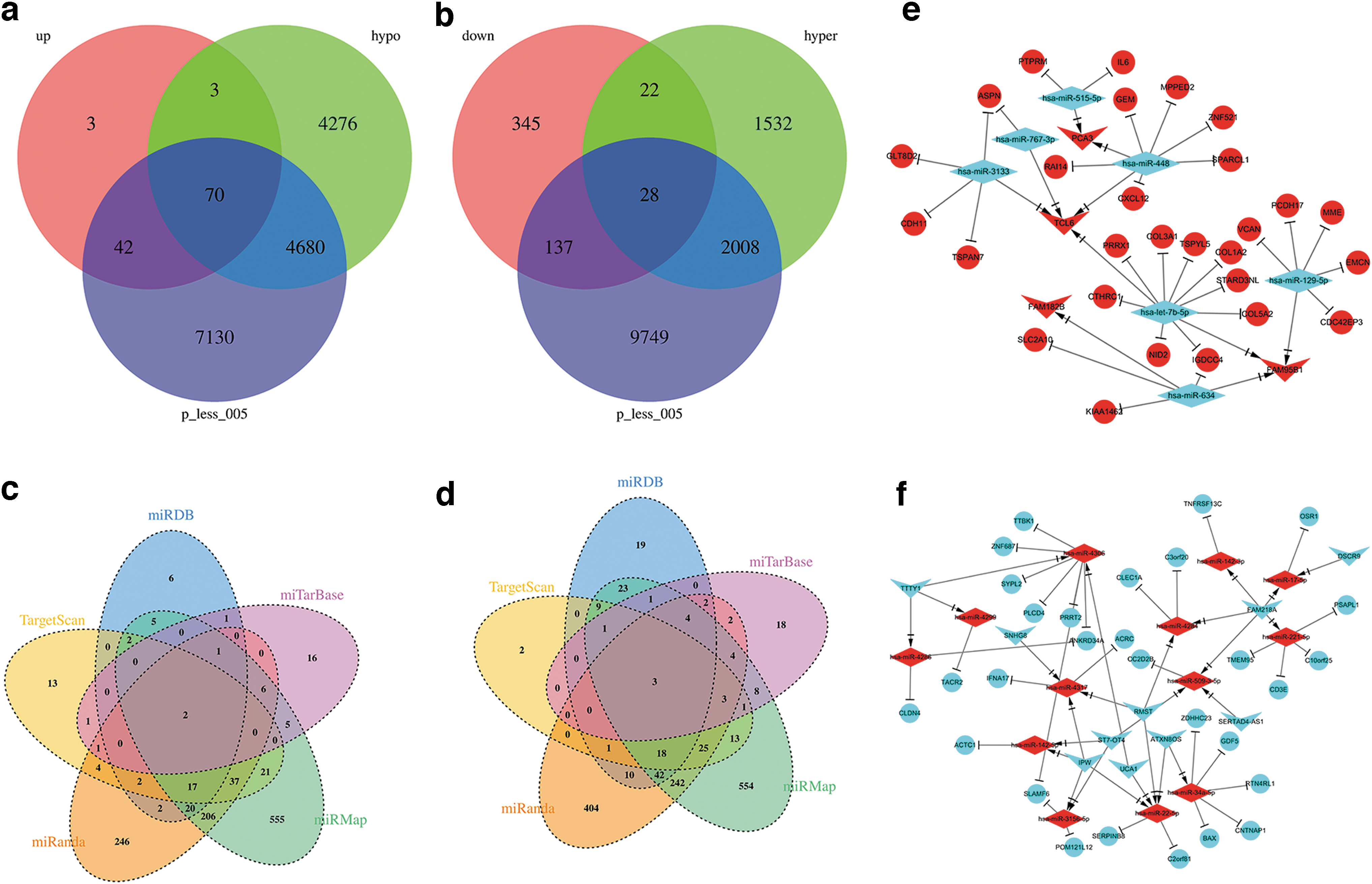
Venn diagrams of methylated mRNA, and Venn diagrams of miRNA-mRNA databases and the lncRNA-miRNA-mRNA network. Venn analysis of
lncRNA, miRNA, and mRNA regulatory networks in periodontitis
The target mRNA of miRNA and the target miRNA or lncRNA was predicted after further screening the results of the coexpression and differential expression analyses (p < 0.05). Through the miRMap, miRanda, miRDB, TargetScan, and miTarBase databases, we predicted the relationship between miRNA-mRNA and miRNA-lncRNA and then constructed a ceRNA network (Fig. 5c-f). The circle, diamond, and inverted triangle nodes represent the mRNA, miRNA, and lncRNA, respectively. Red and blue represent upregulation and downregulation, respectively. The details of the interaction among mRNA, miRNA, and lncRNA are given in Supplementary Tables S1 and S2.
Functional enrichment analysis and hub network construction
To understand the function and mechanism of the 57 mRNAs in the ceRNA network, we used GO and KEGG enrichment in DAVID (q value <0.05 was considered significant). The top 15 GO enrichment terms are given in the bubble chart; these hub genes were mostly enriched in the BP of animal organ development and the generation of neurons (Fig. 6a); the CC of collagen-containing extracellular matrix and intrinsic components of the plasma membrane (Fig. 6b); the MF of extracellular matrix structural constituents conferring tensile strength, calcium ion binding, and cytokine activity (Fig. 6c); and the results of the KEGG analysis (Fig. 6d). Then, select the pathways hsa04064 (nuclear factor κB [NF-κB] signaling pathway) and hsa04620 (Toll-like receptor signaling pathway) related to periodontitis with p < 0.05 from the results of enriched pathways (Supplementary Table S3 and Fig. 6e, f).
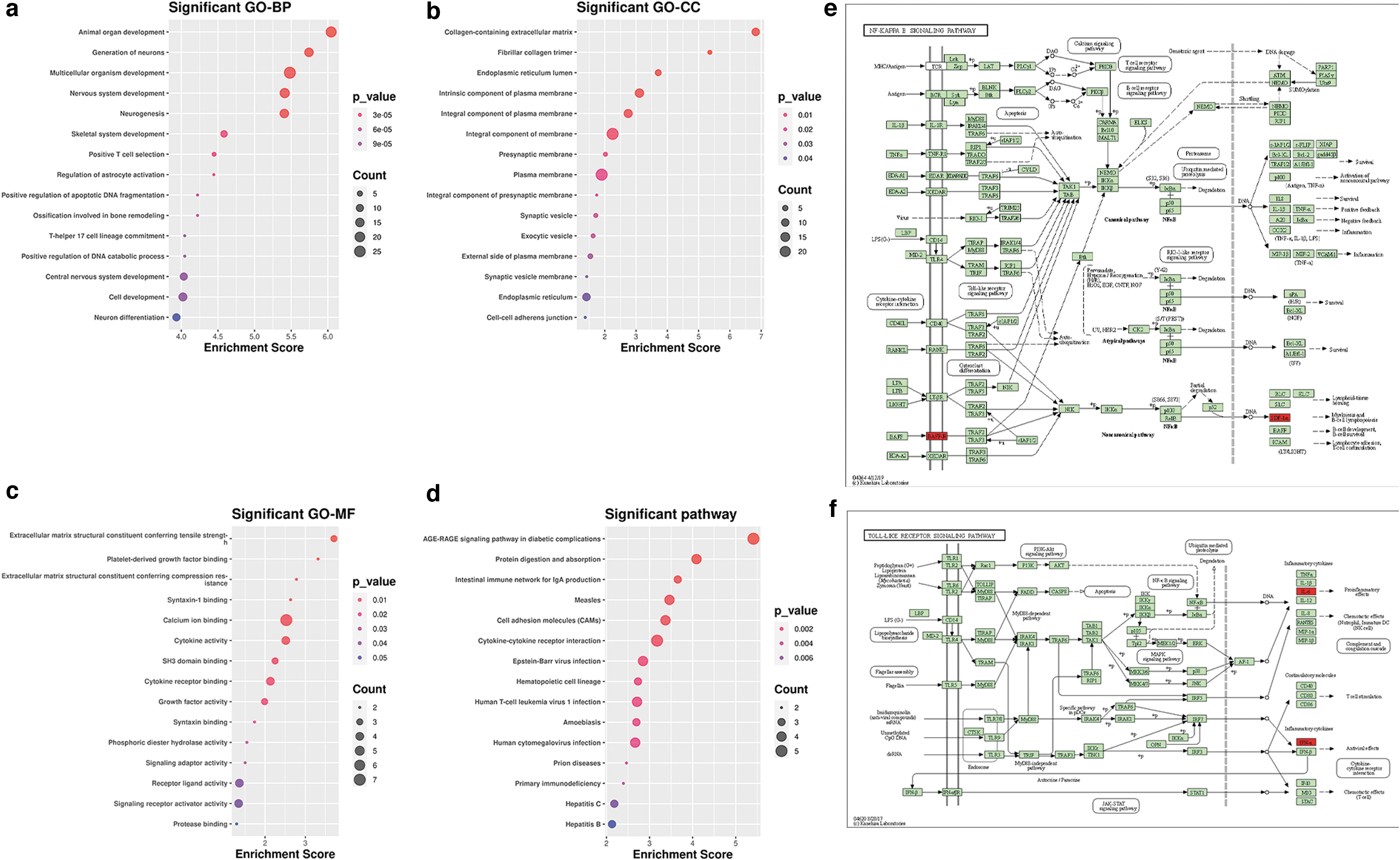
We identified four inflammation-related mRNAs, interleukin 6 (IL-6), interferon alpha 17 (IFNA17), C-X-C motif chemokine ligand 12 (CXCL12), and tumor necrosis factor receptor superfamily member 13C (TNFRSF13C) in the ceRNA network from these two pathways. Based on these four mRNAs, subnetworks were extracted from the ceRNA network results in the previous step (Fig. 7b, c).
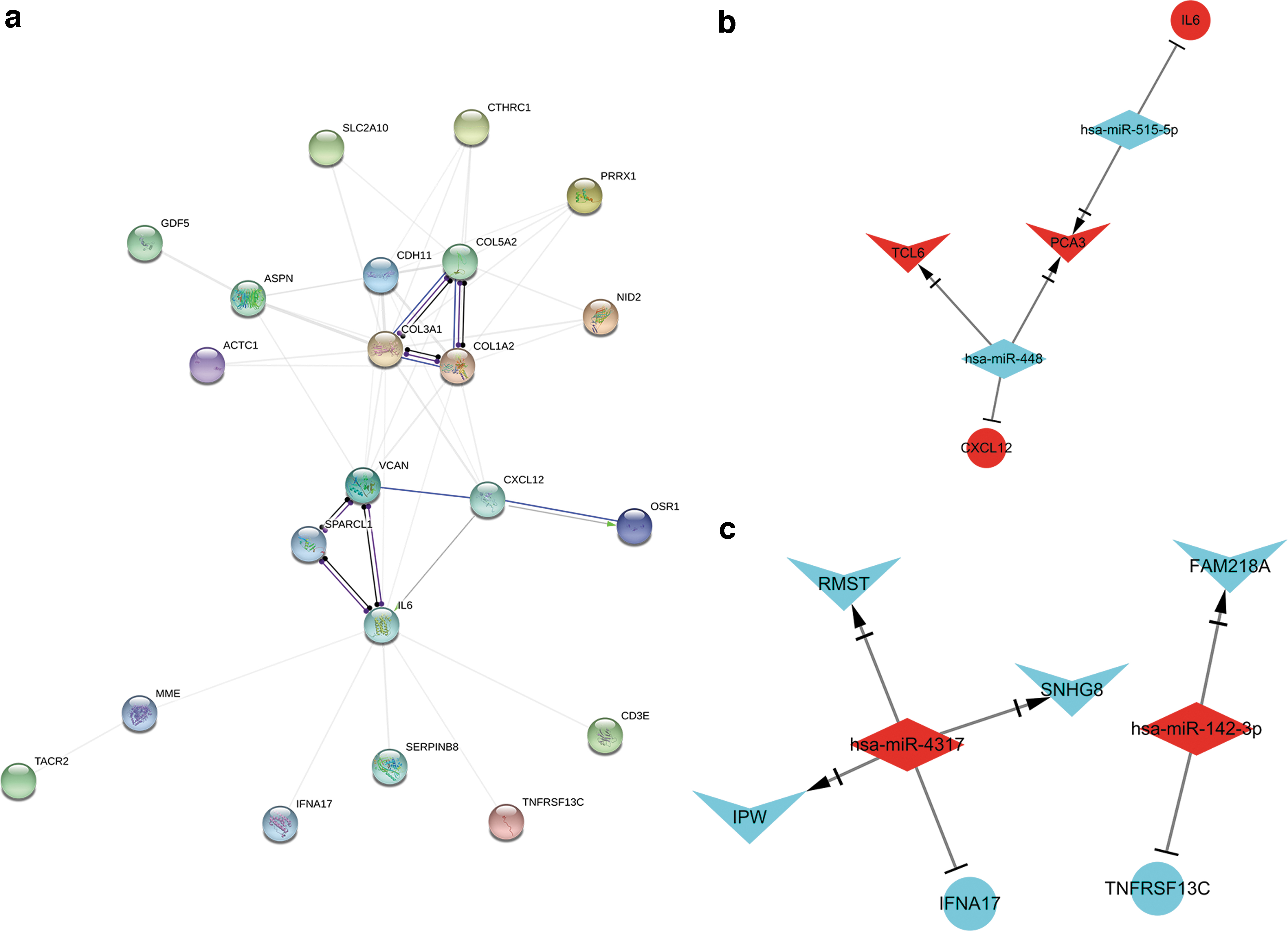
The protein-protein interaction network and the sub-ceRNA network.
PPI network analysis
To assess the interaction of the central genes and identify the key genes, we analyzed the PPI using the STRING online tool and constructed the network using Cytoscape. As given in Figure 7a, the higher the degree of connection, the higher the degree of gene interaction. We selected eight genes with the highest degree of interaction, namely COL3A1, COL1A2, IL-6, COL5A2, CDH11, VCAN, ASPN, and CXCL12. These genes may help to study the biological mechanism of periodontitis and discover potential therapeutic targets.
Discussion
Recently, the rapid development of high-throughput sequencing technologies has allowed researchers to investigate the influence of epigenetic regulatory factors such as DNA methylation and miRNA expression on periodontitis. A new breakthrough has been made in the research direction of the mechanism of periodontitis, and multi-omics research has attracted increasing attention. At present, most studies on the epigenetics of periodontitis have only explored periodontitis-related genes or susceptibility genes regulated by periodontitis-related methylation sites. In this study, the gene expression matrix of periodontitis tissue samples and normal tissue samples in the GEO database was used as the original data, and the modules closely related to periodontitis were selected through the WGCNA method. Subsequent ceRNA network construction and DNA methylation further clarified the genetic and epigenetic mechanisms of periodontitis from two different levels.
The WGCNA algorithm is a classic algorithm for constructing gene coexpression networks. WGCNA is based on high-throughput chip data and assumes that the gene network obeys a scale-free network. By defining a coexpression matrix and adjacency function, and transforming it into a topological matrix, it can identify the gene set modules associated with diseases and consider genes as a whole from biological functions. This method compensates for the shortcomings of traditional methods. By associating clinical information with modules, genes related to clinical features can be further obtained, which is helpful in constructing an expression network of related genes based on the clinical features of disease models.
We downloaded four datasets from the GEO database and performed WGCNA. After further screening through coexpression analysis results and expression difference analysis verification, we predicted the relationship between miRNA and mRNA, and miRNA and lncRNA, and then constructed a ceRNA network. We also analyzed methylated mRNA based on the results of the coexpression analysis. Subsequently, we used mRNA in the ceRNA network for functional enrichment analysis, and selected NF-κB and Toll-like receptors signaling pathways with p < 0.05 and related to periodontitis disease from the pathway enrichment results. From the pathway, four genes including IL6, IFNA17, CXCL12, and TNFRSF13C were found in the ceRNA network.
In previous studies of periodontitis, NF-κB and TLR4 signaling pathways were confirmed to be closely related to periodontitis. First, cytokines involved in inflammation, such as tumor necrosis factor-α (TNF-α) can induce osteoblasts, primary periodontal cells, T cells, and periodontal ligament cells to express the receptor activator of the NF-κB ligand. Combined with receptor activator of NF-κB, it promotes osteoclast production and enhances osteoclast activity to promote alveolar bone resorption (Souto et al., 2014).
Furthermore, studies have shown that TLR4 expression is increased in periodontal ligament cells and fibrocytes in the periodontal tissues of patients with CP. It can be inferred that after the recognition of lipopolysaccharide (LSP) by TLR4 is completed, the signal pathway mediated by it may be closely related to the development of periodontitis (D'souza et al., 2016). Therefore, the hub genes IL-6, IFNA17, CXCL12, and TNFRSF13C in the ceRNA network were identified in the two pathways. These four inflammation-related genes were used to extract subnetworks from the results of the previously constructed ceRNA network, and their importance was determined through the PPI network.
It is well known that cytokines play an important role in host immune defense. Moreover, polymorphisms in cytokine genes can affect the immune response, inflammation, and tissue damage. IL-6 is a cytokine that plays a role in inflammation and B cell maturation. The IL-6 gene is polymorphic in its promoter region (C-G transition at position 174). A previous study reported that the presence of the GG genotype in IL-6-174G/C positively correlated with the risk of peri-implantitis in a study of 103 patients with tissue diseases surrounding implants and proposed that the GG genotype may increase the risk of peri-implantitis (Casado et al., 2013). Nibali et al. (2007) have suggested that IL-6 gene variants may affect the host inflammatory response of periodontal tissues, thereby benefiting the growth of periodontal pathogens. It can be seen that the gene polymorphism of IL-6 may be an important indicator to increase the risk of periodontitis, and it provides a certain clinical reference for the prevention and treatment of periodontitis.
TNFRSF13C, also known as B cell activating factor receptor (BAFF-R), is a membrane protein that recognizes the TNF receptor superfamily of BAFF and participates in the survival and maturation of B cells (Losi et al., 2005). BAFF is an important factor for the activation and maturation of B cells, which can promote the accumulation of B cells in inflammation sites. The gingival tissue in the established period of periodontitis is dominated by plasma cells differentiated from B cells. Studies have shown that the serum BAFF of periodontitis patients is significantly higher than that of healthy people. It is speculated that BAFF can promote the accumulation and maturation of B cells at the site of periodontitis lesions (Nile et al., 2013). Concurrently, some studies have found that BAFF in saliva and serum of periodontitis patients is significantly higher than that in healthy people (Gumus et al., 2014). Therefore, TNFRSF13C is expected to become a new target for early diagnosis or treatment of periodontitis.
CXCL12 is a widely expressed structural chemokine that exerts its function through the CXCR4 receptor. Because CXCL12 can coordinate the transport of a variety of cells expressing CXCR4, CXCL12 plays an important role in many biological functions (Juarez et al., 2004). For a long time, CXCL12 has been considered a typical proinflammatory chemokine because it can attract immune cells to the site of inflammation. However, it has recently been discovered that CXCL12 also has anti-inflammatory effects by mediating the polarization of immune cells toward the anti-inflammatory phenotype (Beider et al., 2014). In recent years, the participation of CXCL12 in the repair and regeneration of periodontitis-damaged tissue has received widespread attention. CXCL12 from different periodontal cells or tissues was confirmed to participate in the regeneration and homeostasis of periodontal tissues by recruiting progenitor cells and stem cells to the injured site (Liu et al., 2015). Based on its clear role in inflammatory diseases and the results of this study, CXCL12 may be a useful biomarker for periodontal disease and its progression (Havens et al., 2008).
lncRNAs participate in almost all normal physiological processes and, by acting as a sponge for miRNA, can affect the expression level of the target mRNA and miRNA. LncRNA PCA3 (prostate cancer antigen 3), previously known as DD3 in prostate cancer biomarkers, was first reported in 1999 (Bussemakers et al., 1999). Human PCA3 is located on chromosome 9q21-22 and consists of four exons. According to reports, PCA3 has regulatory errors in a variety of human diseases, including prostate cancer (Zhang et al., 2019), choriocarcinoma (Wang et al., 2019b), undifferentiated thyroid cancer (Wang et al., 2019a), chronic myeloid leukemia (Sajjadi et al., 2018), and epithelial ovarian cancer (Liu et al., 2017). However, the role of PCA3 in periodontitis is unclear, and there is no relevant research.
T cell leukemia/lymphoma 6 (TCL6), also known as TNG1 or TNG2, is located at the breakpoint cluster area of chromosome 14q32 in T cell leukemia (Saitou et al., 2000). The results of this study indicate that lncRNA TCL6 may be related to periodontitis and has a certain regulatory relationship with miR-448. miR-448 is considered to be one of the miRNAs that are significantly expressed in autoimmune disease animal models and rheumatoid arthritis patients (Jin et al., 2018). Wu et al. (2017) found that miR-448 was deregulated in multiple sclerosis (MS) patients and further promoted the development of MS by inducing Th17 response. Th17 can play a proinflammatory effect by specifically secreting IL-17. IL-17 is a powerful proinflammatory cytokine that binds to receptors and can produce a variety of inflammatory mediators, causing inflammatory cell infiltration and tissue damage. Our previous studies have proved that Th17/IL-17 is involved in the occurrence and development of periodontitis (Chen et al., 2016). Therefore, lncRNA TCL6 is very likely to act as a miR-448 sponge and target CXCL12 to participate in the occurrence and development of periodontitis.
LncRNA rhabdomyosarcoma 2-related transcript (RMST) can regulate neurogenesis (Ng et al., 2013). Decreased RMST has been shown to prevent ischemic brain injury and neuronal damage (Cheng et al., 2020). Recently, some scholars found that downregulation of RMST can improve dopaminergic neuron damage in Parkinson's disease (PD) rats by inhibiting the TLR/NF-κB signaling pathway when they explored the regulation mechanism of RMST in dopaminergic (DA) neuron damage in PD rats (Xu et al., 2017). Of interest, LPS, the main virulence factor of Porphyromonas gingivalis, can induce the expression of MyD88 through TLR4 and the subsequent activation of the NF-κB signaling pathway, resulting in the massive release of inflammatory factors such as TNF-α and IL-6, thereby resulting in the decomposition and destruction of local periodontal tissues and promoting the progression of periodontitis (Molteni et al., 2018; Zhang et al., 2018). Therefore, LncRNA RMST may also regulate the occurrence and development of periodontitis through the TLR/NF-κB signaling pathway.
Our results show that miR-4317 is the target of LncRNA RMST. miR-4317 is a newly discovered miRNA located at 18p11.31. It has been reported to be abnormally expressed in a variety of tumors, such as melanoma (Sand et al., 2013), laryngeal cancer (Janiszewska et al., 2015), and breast cancer (Danza et al., 2014), suggesting that dysregulation of miR-4371 may be involved in the regulation of tumor progression. Although the role of miR-4317 in periodontitis is unclear, the results of this study suggest that LncRNA RMST is likely to act as a miR-4317 sponge and target IFNA17 to participate in the occurrence and development of periodontitis.
DNA methylation is a major epigenetic modification pathway. Our results revealed that BCL2-associated X (BAX) was a downregulated hypermethylated gene; to date, there are no previous reports of its relation to periodontitis. The protein encoded by BAX belongs to the BCL2 protein family, which forms a heterodimer with BCL2 and acts as an apoptosis activator. Recent studies have reported that BAX is associated with T cell acute lymphoblastic leukemia and colorectal cancer.
Among the upregulated hypomethylated genes in this study, C-X-C motif chemokine ligand 2 (CXCL2), a chemokine family member, is noteworthy. A previous study found that CXCL2 can mediate the process by which compressive stress-induced osteoblasts regulate bone formation through the IL-lβ-MyD88 axis (Maeda et al., 2015). However, LPS can induce the expression of MyD88, activate the NF-κB signaling pathway, and release inflammatory factors that promote the progression of periodontitis. Moreover, it has been confirmed that C-X-C motif chemokine ligand 8 (CXCL8) recruits and activates acute inflammatory cells and plays an important role in periodontal disease. From the perspective of epigenetics, CXCL8 is significantly hypomethylated in patients with aggressive periodontitis, suggesting that it is a potential cause of its pathogenesis (Andia et al., 2010). These results suggest that CXCL2 is likely to play an important role in periodontitis.
This study was based on genetics and epigenetics, exploring the pathogenesis and progression mechanism of periodontitis from the levels of ceRNA and methylation, providing new research ideas. However, this study also has certain limitations. First, we used the public dataset in the GEO database; the results need to be verified in a large sample group. Second, the differentially expressed methylation-related genes of the results of this study are rarely found in periodontitis-related research, and there is a lack of corresponding parallel references. More research is needed to confirm the findings. This study sets forth more possibilities for the study of the mechanism of periodontitis and may lead to new therapeutic targets for periodontitis.
Conclusions
We obtained the dataset from the GEO database, screened differentially expressed lncRNA, miRNA, and mRNA, and successfully constructed the ceRNA network. The final key genes (IL-6, IFNA17, CXCL12, and TNFRSF13C) were related to periodontitis. The upregulation of the IL-6 and CXCL12 genes may be independent risk factors for periodontitis. Furthermore, we identified some differentially express methylated genes, such as BAX, which can provide some new insights into the etiology of periodontitis. In addition, CXCL2 and CXCL8 were related to inflammatory pathways. These hub genes could also provide new insights into the etiology of periodontitis.
Footnotes
Acknowledgment
The authors thank all members who participated in the writing of this article.
Authors' Contributions
Y.-Y.F. and C.-X.T. designed this research, Y.-Y.F. and Z.B. performed data analysis, statistical analysis and drafted the article. S.X. assisted in the literature survey. All authors have read the article and agreed to publish it.
Author Disclosure Statement
The authors declare that they have no competing interest.
Funding Information
This research was funded by the National Natural Science Foundation of China (81660185).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.