
Abstract
Background:
Male infertility is a major health concern in couples of childbearing ages. Nonobstructive azoospermia (NOA) is an extreme form of male infertility that affects ∼1% of adult men, and the etiology remains unknown in most cases. Sertoli cell-only syndrome (SCOS) is the most severe type of NOA.
Aims:
To explore novel human candidate variants that cause SCOS.
Methods:
(1) Whole exome sequencing (WES) of 20 men with SCOS, (2) Sanger sequencing of the HELQ gene in an additional 163 men with SCOS, (3) in vitro functional assays, and (4) in vivo studies.
Results:
WES of 20 patients with SCOS led to the identification of two heterozygous missense mutations (M1 and M2) in two unrelated Chinese patients with infertility. Using subsequent Sanger sequencing covering all the coding regions of the HELQ gene for 163 additional SCOS cases, we identified four additional heterozygous mutations (M3-M6) in unrelated patients. In vitro functional analyses revealed that two of these mutations (M5, c.2538T > G and M6, c.2945G > T) might affect the function of the HELQ protein. Two heterozygous mutant mouse models with mutations similar to those of two patients (M5 and M6) did not show any considerable spermatogenic defects.
Conclusion:
Assuming that the mouse models accurately reflect the impact of the mutations, heterozygous HELQ variants alone did not lead to the development of the SCOS phenotype in mice. However, we cannot rule out the risk variants in Chinese or other human populations, and a larger dataset is needed to confirm the association between HELQ mutations with SCOS.
Introduction
Male infertility is a major concern for human reproductive health, affecting ∼7.5% of adult men. Nonobstructive azoospermia (NOA) is a form of male factor infertility affecting ∼1% of adult men, and in the majority of cases, the etiology remains unknown (Tournaye et al., 2017). Sertoli cell-only syndrome (SCOS) is the most severe form of NOA with no germ cells present in the tubules. Several genes involved in spermatogenesis, such as ETV5 (O'Bryan et al., 2012), SEPTIN12 (Miyakawa et al., 2012), NANOS1 (Kusz-Zamelczyk et al., 2013), LRWD1 (Miyamoto et al., 2014), PLK4 (Miyamoto et al., 2016), FANCA (Krausz et al., 2019), FANCM (Kasak et al., 2018), and USP26 (Arafat et al., 2020), have been reported to cause SCOS. However, many idiopathic SCOS cases may be ascribed to genetic defects that have not yet been identified (Hotaling and Carrell, 2014). Genes involved in spermatogonia stem cell proliferation and DNA interstrand crosslink repair play important roles in the maintenance of germ cells. Hence, these genes should be given special attention in SCOS cases, which generally manifest as loss of germ cells (Krausz et al., 2019).
HELQ is a DNA helicase required for DNA interstrand crosslink repair through its association with RAD51 paralogs to prevent germ cell loss and cancer predisposition (Adelman et al., 2013). HELQ is expressed in several human tissues, with the strongest expression observed in the testes, followed by the skeletal muscle, heart, and ovaries (Marini et al., 2003). In mice, genetic ablation of Helq results in subfertility, germ cell loss, ICL sensitivity, and tumor predisposition, whereas Helq heterozygous mice show haplo-insufficiency (Adelman et al., 2013). Whether mutations in HELQ can cause male infertility in humans is unknown.
In the current study, whole-exome sequencing (WES) and direct Sanger sequencing identified six heterozygous mutations in eight unrelated patients with SCOS. In vitro functional analysis revealed that the function of the HELQ protein might be affected by two variants. However, the two heterozygous mutant mouse models did not show any subtle spermatogenic defects.
Materials and Methods
Study population
Testicular biopsy tissues were obtained from azoospermic patients who were referred to in vitro fertilization clinics because of the absence of sperm in their ejaculate after meticulous search of spermatozoa. Biopsy specimens were processed for Hematoxylin and Eosin (H&E) staining and histologically diagnosed as NOA. Peripheral blood samples were collected from all participants for genetic evaluation, including karyotyping and Y-chromosome microdeletions. Patients with a history of acquired infertility were excluded, and only those who had a normal 46, XY karyotype, none of whom had Y-chromosome microdeletion, varicocele, cryptorchidism, or any history of chemotherapy/radiotherapy treatment, were considered. A total of 183 histologically confirmed patients with SCOS who met our inclusion criteria were selected. All the studies on patients were performed after getting informed consent and this project was approved by University of Science and Technology of China (USTC) USTCEC202000003.
Whole-exome sequencing
DNA extraction and WES were performed as described previously (Yin et al., 2019). The variants were filtered based on the following steps (Supplementary Fig. S1): (1) variants that could potentially affect the protein sequence (frameshift substitution, stop-gain, splicing, nonsynonymous variants) were retained; (2) variants that had genomic frequency MAF >0.01 in human genetic variation databases (1000 Genomes, ESP6500, or ExAC), and variants present in our in-house WES database generated from 578 fertile males (254 Chinese, 41 Pakistani, and 283 European) were omitted; (3) variants in genes that may be functional in spermatogenesis predicted using SpermatogenesisOnline (Zhang et al., 2013) and literature were retained; (4) variants reported to be involved in proliferation of spermatogonia or required to prevent germ cell loss were retained; (5) variants predicted to be deleterious by more than half of the seven software covering them were chosen as candidates. The list of software used for deleterious prediction is given in Supplementary Table S1, and their categorical prediction values are listed in Supplementary Table S2.
Sanger sequencing of HELQ coding regions
Direct Sanger sequencing of HELQ coding regions was performed on polymerase chain reaction (PCR) fragments amplified using genomic DNA from peripheral blood cells and gene-specific primers. The same seven bioinformatics tools, SIFT, PolyPhen2, Mutation Taster, Mutation assessor, SiPhy, GERP++, and fathmm were used to predict noxious effects of HELQ variants in the patients. The primers used are shown in Supplementary Table S3.
In vitro functional analysis
Wild-type and mutant HELQ (GFP-tagged) proteins were expressed in HEK293T cells to analyze the expression and localization of the HELQ protein. The HELQ knockout (KO) U2OS cell line was generated using the CRISPR/Cas9 genome editing technique. HELQ wild-type and mutant vectors were constructed and transfected into HELQ KO U2OS cells. After 24 h of transfection, 2 μM mitomycin C (MMC) was added to the culture medium. The cells were incubated, fixed, and stained for the survival assays (Supplementary Fig. S2). Relevant primers are available upon request.
KI mice models
Two heterozygous HELQ mutant (M5, WT/p.Y804* and M6, Helq WT/p.C940F) mice carrying mutations similar to those of our two patients were generated using CRISPR-Cas9 technology, as previously described (Zhang et al., 2020). All in vivo studies were performed using C57BL/6 mice. Testes and epididymides were collected from 60-day-old WT and KI mice and used for gross morphological examination, sperm count, and H&E staining.
Results
Identification of heterozygous HELQ variants in men with NOA
To understand the underlying genetic causes of NOA, we performed WES on 20 patients with SCOS. The WES data were analyzed using an improved version of our previously described filtration method (Yin et al., 2019). We selected all variants that followed the filtration criteria (Supplementary Fig. S1). The variants that could potentially affect the protein sequence (frameshift substitution, stop-gain, splicing, and nonsynonymous variants) were chosen. Variants with genomic frequency MAF >0.01 in human genetic variation databases (1000 Genomes, ESP6500, or ExAC) and variants present in our in-house WES database generated from 578 fertile males (254 Chinese, 41 Pakistani, and 283 European) were omitted. Variants in genes that may be functional in spermatogenesis predicted using SpermatogenesisOnline (Zhang et al., 2013) and literature were kept. Nonsynonymous variants predicted to be deleterious by more than half of the seven software covering them were taken as candidate variants. In this study, variants in known genes related to SCOS or novel candidate genes that were functionally related to germ cell maintenance, but had not yet been reported as SCOS-causative genes, were preferred. Two heterozygous HELQ mutations (M1, c.47A > G and M2, c.1468A > G) were identified in two unrelated patients with NOA. Moreover, no other rare variants in known SCOS-causative or candidate genes (Supplementary Table S4) were observed in these two patients.
Furthermore, to explore the association of HELQ mutations with SCOS, we performed direct screening of HELQ in another group of 163 patients with SCOS. Sanger sequencing of all the coding regions of HELQ led to the identification of four additional novel heterozygous mutations, including one nonsense (M5, c.2538T>G) and three nonsynonymous missense mutations (M3, c.368T > A, M4, c.1567G > A, and M6, c.2945G > T) in four unrelated patients (Fig. 1a). Moreover, M2 and M5 were also found in two unrelated patients with SCOS. All the variants, predicted to be noxious by more than half of the computational tools covered in this study, had extremely low frequencies in the human population, were evolutionarily conserved across many mammalian species (Fig. 1b), and none was found in our in-house normal controls (Table 1).
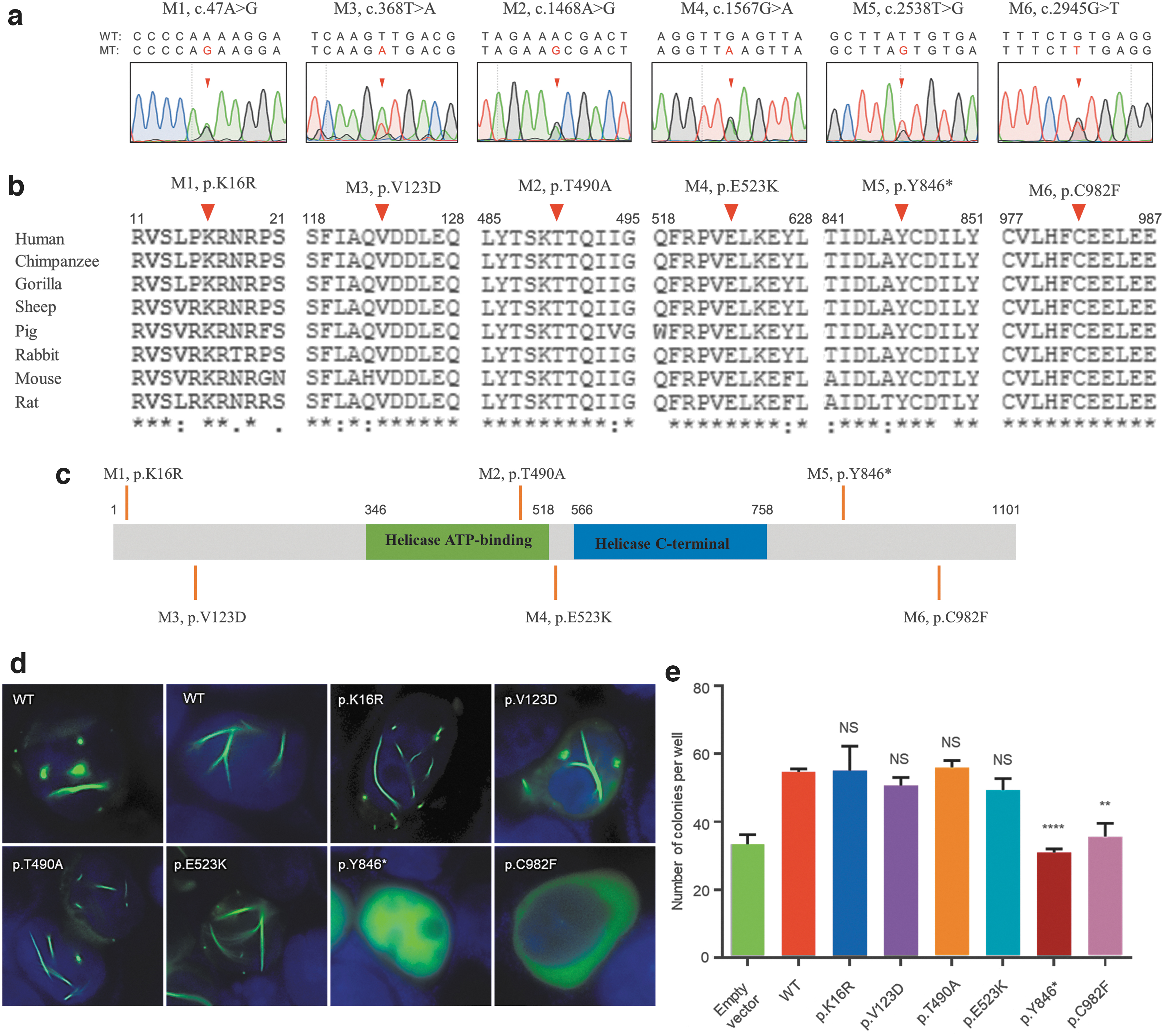
HELQ heterozygous variants identified in NOA patients with SCOS.
Overview of HELQ Heterozygous Variants Identified in Infertile Patients with Sertoli Cell-Only Syndrome
In-house 578 fertile men, including 254 Chinese, 41 Pakistani, and 283 Europeans.
Ratio of the number of in silico programs predicting deleteriousness by the number of programs covering the mutation. Following in silico programs that were used: SIFT, PolyPhen2, Mutation taster, Mutation assessor, SiPhy, GERP++, and fathmm. Individual Categorical prediction values of used software are written elsewhere in Supplementary Table S3.
SCOS, Sertoli cell-only syndrome.
In vitro functional assays of HELQ variants
To examine the impact of the identified HELQ variants, we performed in vitro functional assays. First, the putative effects of these mutations on HELQ protein expression and localization were investigated. The mutant and wild-type HELQ (GFP-tagged) proteins were expressed in human HEK293T cells. As expected, wild-type HELQ protein was detected within the nucleus in filamentous form, but the two mutant proteins (p.Y846* and p.C982F) were found in an altered form as compared with the wild-type protein (Fig. 1d). Furthermore, the HELQ protein also exhibited abnormal intracellular localization in the p.C982F mutant. However, the other four mutants (p.K16R, p.V123D, p.T490A, and p.E523K) displayed normal filamentous protein structure and localization similar to those observed in the wild type.
Second, to determine whether these mutations affected the function of HELQ protein, clonogenic survival assays were performed in U2OS cells exposed to MMC. Two mutant colonies (p.Y846* and p.C982F) showed higher sensitivity to MMC than wild-type cells, suggesting that the function of HELQ as ICL resistance might have been affected in these two mutant colonies (Fig. 1e).
Heterozygous KI mice manifested normal spermatogenesis
To understand whether these two heterozygous mutations found in our patients can indeed cause spermatogenic defects, we generated two heterozygous (Helq+/M5 and Helq+/M6) mutant mouse models carrying mutations similar to those of our two patients. After confirmation of mutations using Sanger sequencing, testes of 60-day-old WT and each mutant mouse were examined for gross morphology. Both mutant mice showed normal testicular size (Fig. 2a, b). and testis-to-body weight ratio similar to that in wild-type mice (Fig. 2c). Consistent with these observations, both KI mice exhibited no marked difference in cauda epididymal sperm count when compared with wild-type mice (Fig. 2d). Further histological analysis was performed to determine whether these heterozygous mutations caused any subclinical effects on spermatogenesis. The seminiferous tubules from both types of mutant mice showed normal spermatogenesis and all spermatogenic stages were similar to those observed in the wild-type mice (Fig. 2e).
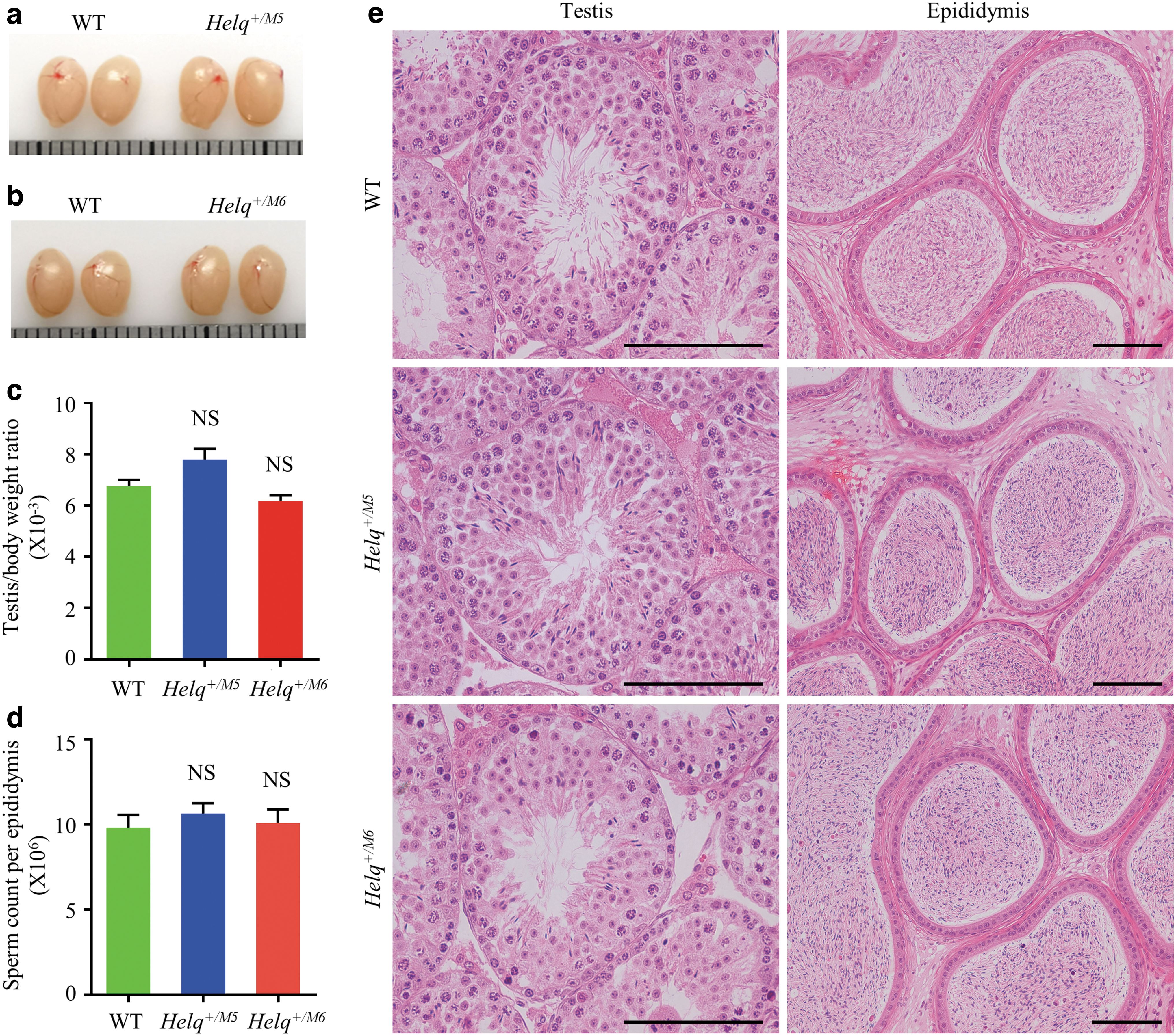
Spermatogenesis of Helq+/M5 and Helq+/M6 mice.
Discussion
Here, we studied the genetic causes of male infertility in NOA with testicular histology of SCOS as part of our project to develop a noninvasive diagnosis for patients with SCOS. WES analysis of 20 patients led to the identification of two novel heterozygous variants in the HELQ gene, while direct Sanger sequencing of the HELQ gene in another group of 163 patients with SCOS allowed the identification of four additional novel heterozygous variants. In vitro functional analysis suggested that two of these mutants could affect the structure and function of the HELQ protein. However, two heterozygous mutant mouse models (Helq+/M5 and Helq+/M6) carrying heterozygous mutations similar to those of our two patients did not show any spermatogenic defects. A previous study in mice showed that genetic ablation of Helq resulted in subfertility, germ cell loss, ICL sensitivity, and tumor predisposition (Adelman et al., 2013). Hence, we cannot rule out HELQ as a risk variant in Chinese or other populations, and a larger dataset is needed to confirm the association between HELQ mutations and SCOS.
Several studies on individuals with sporadic infertility have identified variants using a number of approaches for sequencing the putative gene, suggesting the association of suspected variants with infertility if the variant is unique to patients with infertility and predicted noxious using bioinformatics tools. In the future era of personalized medicine, however, validation of causative alleles would remain challenging as putative infertility alleles of human SPATA16 (R238Q), DMC1 (M200V), and MND1 (K85M) were shown to have no impact on fertility or spermatogenesis in mice (Fujihara et al., 2017; Tran and Schimenti 2018; Tran et al., 2019). Another study revealed that only 25% of highly damaging predicted nonsynonymous SNPs in the human population produced an infertility-related phenotype when modeled in mice (Singh and Schimenti, 2015). This study also has some limitations: the sample size was not determined before the onset of the study and the number of enrolled patients was not sufficient to draw a definite conclusion.
In summary, assuming that mouse models are more physiologically relevant to reflect the impact of the putative infertility allele, these heterozygous variants do not cause the SCOS phenotype in mice alone. This study emphasized the importance of in vivo functional studies to provide evidence for the consequences of putative polymorphisms and de novo mutations associated with sporadic infertility before taking clinical action.
Ethics Approval
All the experiments and examination of laboratory animals were conducted according to the institutional rules of the Institutional Animal Care Committee of the University of Science and Technology of China.
Informed Consent
Informed consent was obtained from all participants included in the study.
Footnotes
Authors' Contributions
Q.S., G.M., and L.Y. conceived and designed the experiments. L.Y. and G.M. performed the experiments. R.K. and L.Y. analyzed the data. G.M. and Z.H. wrote the article. I.U., I.K., M.K., Z.H., R.K., and Q.S. modified the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Key Research and Developmental Program of China (2016YFC1000600 and 2018YFC1004700), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB19000000), the National Natural Science Foundation of China (31890780, 31630050, and 31871514), Major Program of Development Foundation of Hefei Center for Physical Science and Technology (2018ZYFX005), and the Fundamental Research Funds for the Central Universities (YD2070002006).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.