
Abstract
Aims:
Autosomal recessive primary microcephaly (MCPH) is a clinically rare and genetically highly heterogeneous developmental disorder. Biallelic variants in the abnormal spindle-like microcephaly-associated (ASPM) gene account for 40% to 68% of all MCPH cases. This study was designed to elucidate the genetic basis of MCPH in an extended family. To highlight recurrent mutations useful in implementing genetic testing programs, we further aimed to carry out a descriptive review of the reported ASPM mutations.
Materials and Methods:
A large inbred kindred with seven affected members was investigated, and detailed clinical and behavioral assessments were carried out. Single nucleotide polymorphism (SNP)-based homozygosity mapping and exome sequencing were performed.
Results:
Affected individuals had characteristic features, including small head, receding forehead, mild to moderate intellectual disability, developmental delay, short stature, apraxia, and behavioral anomalies. We mapped the disease gene locus and detected a rare frameshift deletion c.6854_6855del (p.(Leu2285GlnfsTer32)) in exon 18 of ASPM. A total of 215 mutations in ASPM have been reported in at least 453 families, nearly 50% of which are of Pakistani origin. These mutations can be classified as recurrent, founder or private in Pakistani and other populations.
Conclusion:
SNP-based homozygosity mapping and exome sequencing are essential in delineating the genetically distinct microcephaly types. The highlighted recurrent mutations in ASPM could be useful in implementing genetic testing programs for MCPH.
Introduction
Autosomal recessive primary microcephaly (MCPH; MIM-251200) is a clinically and genetically highly heterogeneous developmental anomaly that is characterized by prenatal onset of brain growth impairment resulting in reduced brain volume, which is measured as an occipitofrontal circumference equal to or >2 standard deviations (SDs) and 3 SDs below the age- and sex-matched means at birth and 6 months, respectively. This condition leads to intellectual disability (ID) without any significant neurological deficit (Barbelanne and Tsang, 2014; Von der Hagen et al., 2014). At least 28 loci are known for MCPH, and the corresponding genes have been discovered (https://www.omim.org/phenotypicSeries/PS251200).
MCPH1-17, MCPH19-25, and MCPH28 are inherited in an autosomal recessive manner, whereas MCPH18, MCPH26, and MCPH27 are autosomal dominant. Biallelic variants in abnormal spindle-like microcephaly-associated (ASPM) gene (MIM-605481) are the most frequent cause of MCPH and account for 40-68% of primary microcephaly cases (Zaqout et al., 2017; Letard et al., 2018). Nearly half of all reported MCPH patients originate from Pakistan.
Genetically distinct microcephaly types share high phenotypic similarity such as congenital onset, short stature, mild to severe ID, receding forehead, decreased brain weight, disproportionately thin cerebral cortex, developmental and speech delay, and speech apraxia (OMIM; Faheem et al., 2015; Shaheen et al., 2019; Jean et al., 2020). High-throughput methods such as single nucleotide polymorphism (SNP)-based genotyping and exome sequencing are essential to delineate genetically heterogeneous conditions like microcephaly.
We present a Pakistani kindred with seven microcephalic members. Through SNP-based genotyping and homozygosity mapping followed by exome sequencing, we showed that a rare variant in ASPM segregates with the malformation in the family. We also carried out a descriptive review of the reported ASPM mutations and highlight recurrent mutations useful in implementing genetic testing programs.
Materials and Methods
Sample collection and clinical investigations
Peripheral blood samples were collected after obtaining informed consent according to the Declaration of Helsinki II. The study protocol was approved by the Ethics Review Committee of Quaid-i-Azam University (DAS-1070) and the Istanbul Technical University Ethics Review Board (MBG.22/2014).
The family is from a remote area of Punjab, Pakistan. The five-generation pedigree strongly suggested an autosomal recessive pattern (Fig. 1A). Seven family members (three males and four females) at ages 16 to 32 years were affected. In total 10 family members (5 affected and 5 unaffected) were physically examined with the help of local physicians. Photographs of all participants were taken, and anthropometric measurements of affected subjects were obtained.
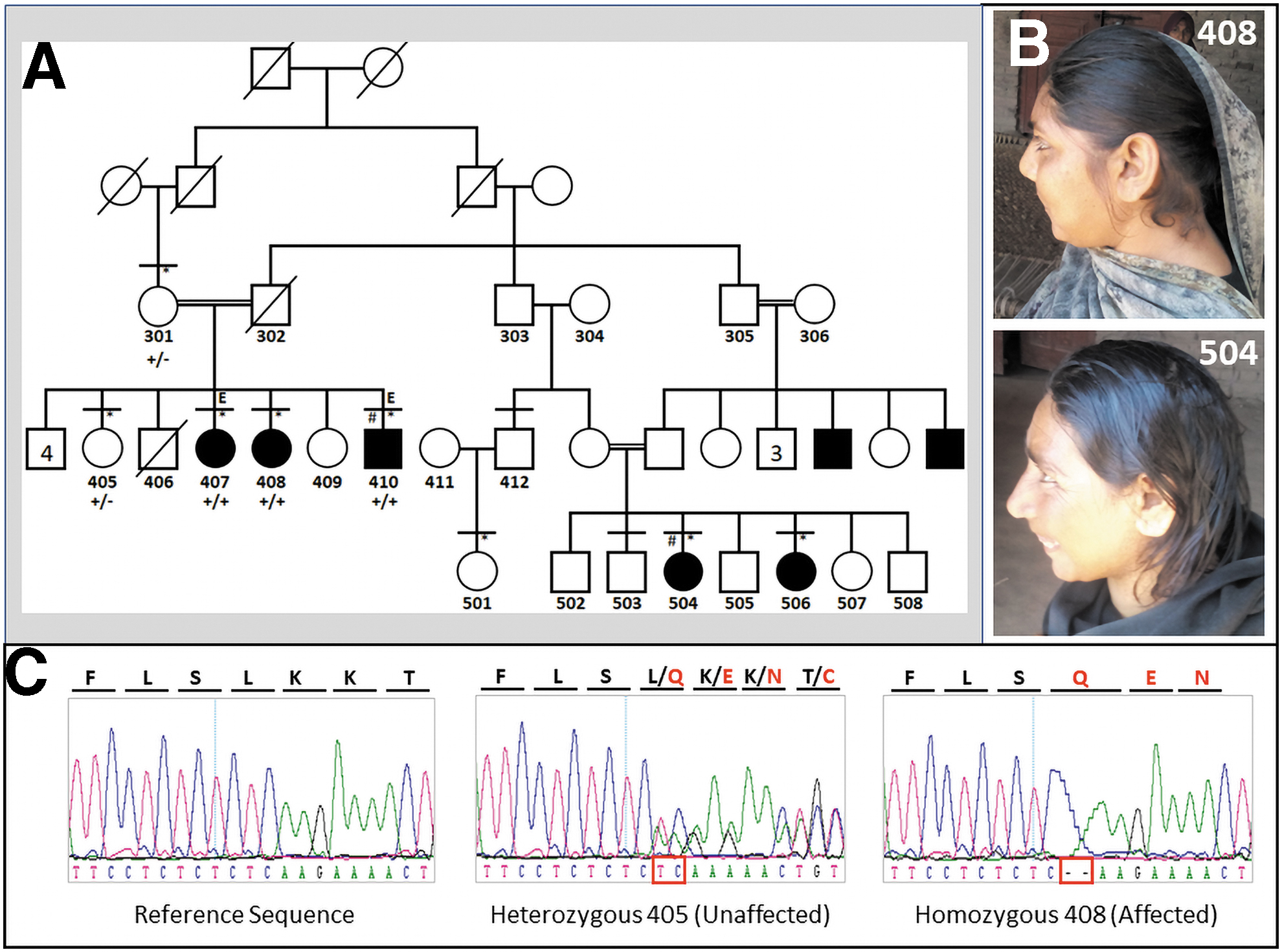
Family and patients.
Genetic analyses
Genotype data for 710K SNP markers for a mixture of DNA samples of two affected individuals (410 and 504) were generated using Illumina Human OmniExpress-24 BeadChip. Intervals >1 Mb with shared homozygosity were detected through Homozygosity Mapper and manual inspection on MS-Excel.
DNA sample of affected sibs 407 and 410 were subjected to exome sequencing with Illumina TruSeq Exome Capture kit followed by massively parallel paired-end sequencing with Illumina HiSeq 2000 (Illumina, San Diego, CA). Variant calling and annotation of exome data were performed by using BWA, SAMTools, and ANNOVAR (2019Oct24). The alignments were visualized with BamView. The threshold for sufficient coverage was assumed as four reads, and the minimum accepted threshold for quality score was 40.
In candidate regions, rare (frequency <0.01) and novel variants were selected according to the information in public databases gnomAD (v2.1.1) that contains thousands of Pakistani exomes, 1000 Genome and ESP6500 (SI-V2), and those possibly homozygous (alternate depths >0.60) and affecting protein structure were considered. Variants found in in-laboratory exome files were excluded. Sanger sequencing was carried out to assess the segregation of the variant with the disease. The genome assembly of hg19 was utilized.
Results
Clinical findings
Patients have small heads, mild to moderate ID, short stature, narrow and oval shaped faces, receding foreheads, large ears, and prominent noses (Table 1 and Fig. 1B). They have attention deficit behavior and speech apraxia. They never attended a school of any kind, because of low ID. Affected males spend time wandering in the streets and have no concept of money. They are not able to perform any conceptual work but recognize relatives and find their way home. They have sense of self-respect. They are comfortable with strangers and are friendly towards them. For instance, female individual 504 at age 34 always has a smiley face. According to family elders, affected relatives had delayed developmental landmarks, including delayed crawling, walking, and toilet training.
Physical and Clinical Features of Affected Family Members
As per criteria of the American Psychiatric Association. (2013).
+, feature present; −, feature absent.
ID, intellectual disability.
They do not have hyperactive or aggressive behavior, and epilepsy and bipolar episodes were not observed. Anthropometric measurements showed marked developmental failures (Supplementary Table S1).
Genetic findings
SNP genotypes were used to detect 22 autosomal homozygous regions >1 Mb shared by the two affected siblings. Those regions were scrutinized for potential candidate genes through GeneDistiller. Exome filtration strategy revealed a total of eight variants common to the exome files of the two affected sibs (Supplementary Table S2). Seven of those variants were functionally not relevant to the phenotype and did not fall in a homozygous interval. Frameshift variant c.6854_6855del (p.(Leu2285GlnfsTer32); NM_018136.4) in ASPM exon 18 was detected in a shared homozygosity region of 7.83 Mb at 1q31. It is extremely rare, with allele frequency 0.00003268 (1 allele in 30604 alleles from South Asian samples in gnomAD). It has been reported in compound heterozygous in an MCPH family (Rasool et al., 2020).
In the exome file, no pathogenic variant was found in other MCPH-related genes.
Descriptive summary of ASPM mutations
To investigate the recurrence and to understand the pattern of mutation, we compiled all ASPM variants assembled by Letard et al., (2018) and Rasool et al., (2020) and added more recently reported variants (Batool et al., 2021; Khan et al., 2021; Naseer et al., 2021). To investigate recurrence and detect potential mutational hotspots, data were extracted from published reports using large microcephaly cohorts (Tan et al., 2014; Ahmad et al., 2017; Wang et al., 2017; Ahmed et al., 2019; Shaheen et al., 2019). Those data extend the number of different disease variants to 215 in at least 453 families.
Reported causative mutations are dispersed throughout the gene. A summary of the distribution is presented in Figure 2. There is a direct relationship between exon length and the number of variants (Spearman correlation R2 = 0.9771), indicating that there is no mutation hotspot. The majority (n = 200) of the mutations fall across coding regions; only 15 are intronic. There are two complex rearrangements and two large deletions that encompass several exons/introns.
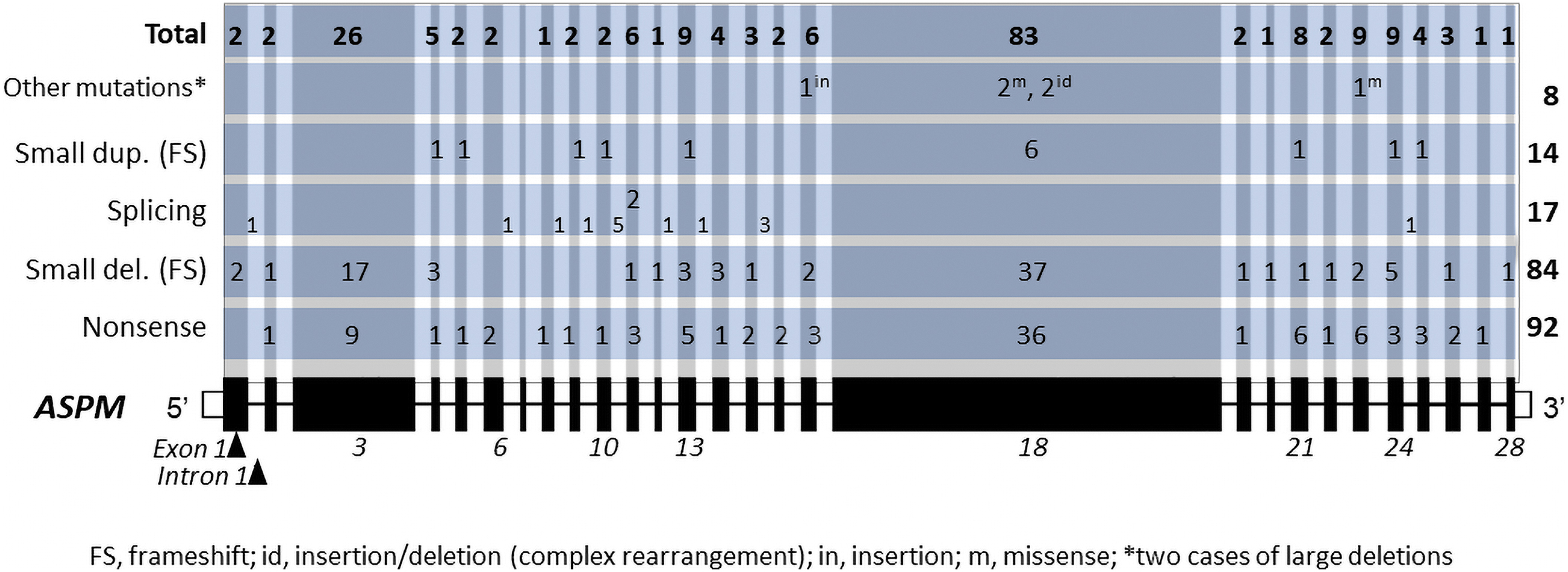
Distribution of known MCPH causing ASPM variants. MCPH, autosomal recessive primary microcephaly.
Majority of the mutations are nonsense (n = 92), followed by small frameshift deletions (n = 84), splicing (n = 17), and small frameshift duplications (n = 14) (Table 2). Missense variants are scarce (n = 3), raising the question whether mild mutations are tolerated. Nonsense mutations are highest in exon 18 (n = 36), followed by exon 13 (n = 9). Small frameshift deletions are also more prevalent in exon 18 and 13. Highest number of splicing variants is in intron 10. Exon 7 is the smallest exon (68 bp) and has no known mutation.
Abnormal Spindle-Like Microcephaly-Associated Mutation Types, Number of Affected Families, and the Origin of the Families
FS, frameshift.
Microcephaly families of Pakistani origin have the highest contribution to the mutational heterogeneity of ASPM. Table 3 shows the recurrent mutations and the number of reported families for mutation types. The most common variant is c.3978G>A (p.(Trp1326Ter)) followed by c.7782_7783delGA (p.(Lys2595SerfsTer6)), occurring in at least 77 and 21 families, respectively (totally 22% of all families). At least seven mutations are likely founder mutations reported primarily or exclusively in the Pakistani population. Nearly half of the mutations (n = 149) are private, with 30% contribution of novel mutations from Pakistani patients.
Recurrent Mutations and Number of Families with the Variant
FS, frameshift.
Among at least 110 base substitutions, the C > T transition is the most frequent variant (n = 51) followed by transversion G > A (n = 15), both likely due to deamination of a C. The small frameshift deletions and duplications comprising 98 of the variants could be due to DNA polymerase slippage.
Discussion
In this study, we demonstrate that homozygous frameshift deletion c.6854_6855del (p.(Leu2285GlnfsTer32)) underlies MCPH microcephaly in a large Pakistani kindred. Due to high genetic heterogeneity in MCPH, we initially launched SNP-based homozygosity mapping and detected homozygous intervals shared between two affected relatives in different branches of the kindred. SNP genotyping coupled with exome sequencing ruled out linkage to any other known recessive MCPH locus. The two-base pair deletion detected in the present family falls in the IQ repeats of ASPM and is deduced to cause a shift in the translational frame and incorporation of 32 non-native amino acids before termination, or nonsense-mediated RNA decay (NMD) could occur prior to translation.
The resulting truncation causes the deletion of ∼33% of the native protein. Functional studies on ASPM have revealed that calmodulin binding to IQ motifs induces a conformational change in proteins that regulate the binding of actin to the amino terminal CH domains. It has been proposed that changes in ASPM alter the orientation of the mitotic spindle of neuroblasts, which induces symmetric mitosis that results in two progenitor cells (Kouprina et al., 2004). IQ motifs have been suggested to play an essential role in determining brain size (Zhang, 2003).
ASPM is a large gene with 28 exons and codes for a 3477-amino acid protein. Majority of the reported mutations are nonsense, small deletions, splicing, or small duplications. Those mutations are spread throughout the protein, and the majority is predicted to generate either a premature truncated protein or unstable RNA that is degraded by NMD (Abdel-Hamid et al., 2016; Letard et al., 2018). In summary, the majority of ASPM mutations likely cause loss-of-function of the encoded protein (Letard et al., 2018). Based on the accumulated data on recurrent and founder mutations, it should be possible to implement genetic testing and molecular diagnosis of MCPH. Furthermore, as remarked by Letard et al. (2018), functional studies are warranted to prove the pathogenicity of the reported variants.
Previously, it was observed that there was no straightforward genotype-phenotype correlation between mutation type or site and the head size and other clinical features associated with MCPH (Nicholas et al., 2009; Passemard et al., 2009). It is, however, pertinent to mention that the genotype-phenotype studies are limited due to unavailability of detailed clinical description, quantitative evaluations of cognitive, neurodevelopmental and behavioral variables, and neuroimaging studies. It also remains to be elucidated whether the functional and phenotypic impact of frameshift and nonsense mutations occurring in the initial exons is milder than such mutations occurring at the end of gene, or vice versa. Furthermore, the consequences of the mutations falling in four distinguishable regions of ASPM protein remain unknown.
ASPM is the human ortholog of the Drosophila melanogaster “abnormal spindle” gene (asp), which has a pivotal role in normal mitotic spindle function in embryonic neuroblasts. In mouse, it has been shown that Aspm protein has a role in the regulation of mitotic spindle and neurogenesis (Fish et al., 2006). Mutations in Aspm are also known to cause reduction in the size of brain in mice (Pulvers et al., 2010). These evidences are suggestive that mutations in ASPM impair the development of brain by perturbing the orientation of the mitotic spindle and can decrease the number of neuronal cells by affecting the asymmetrical to symmetrical cell division ratios (Thornton and Woods, 2009). Further molecular studies are required to understand the detailed cellular mechanisms involved in the pathogenicity of ASPM defects in microcephaly.
Conclusions
SNP-based homozygosity mapping and exome sequencing are essential in delineating the genetically distinct microcephaly types. The mutation spectrum of ASPM comprises recurrent, founder, and private mutations, some of which have been reported primarily or exclusively in the Pakistani population. The highlighted recurrent mutations in ASPM could be useful in implementing genetic testing programs for MCPH.
Footnotes
Authors' Contributions
All persons who meet authorship criteria are listed as authors.
Acknowledgments
We thank family members for their cooperation.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by URF-QAU, Pakistan [grant number DFBS/2014/3230, 2013-2014] and the Scientific and Technological Research Council of Turkey [TÜBITAK, grant number 114Z829].
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.