
Abstract
Purpose:
To explore the genetic defects in two Chinese families with X-linked Norrie disease (ND).
Methods:
We analyzed two Chinese families with ND at molecular level through clinical exome sequencing and the variations were identified by Sanger sequencing.
Results:
Two genetic variations were found in the NDP gene by clinical exome sequencing, a partial deletion of 801 bp contained the whole exon 2 and a missense variant (164G>A) within codon 55 that resulted in the interchange of cysteine by phenylalanine. Clinical findings were more severe in the patients who presented the missense variant.
Conclusion:
We report two genetic variations in the NDP gene in Chinese that extend the mutational and phenotypic spectra of NDP gene, and also demonstrate the feasibility of clinical exome sequencing in application of molecular diagnosis.
Introduction
Norrie disease (ND) (MIM no. 310600) is a rare X-linked recessive disorder characterized by congenital blindness with a prevalence of 1 per 100,000 people, among whom 30-50% of these patients have varying degrees of neurodevelopmental retardation and one-third develop progressive sensorineural deafness (Riveiro-Alvarez et al, 2005; Rodriguez-Munoz et al, 2018). In ND patients, retinal malformations occur early during embryogenesis and the most prominent feature in newborn is an intraocular mass that can be misdiagnosed and ultimately leads to ocular shrinkage (Chen et al, 1992).
The Norrie disease protein (NDP) gene has been found to be the pathogenic basis of ND. The NDP gene is located on chromosome Xp11.3 and first identified by two groups (Berger et al, 1992; Chen et al, 1992). The NDP-encoded secretory protein, consisting of 133 amino acids, is widely expressed in different tissues, such as the eye, ear, and brain (Chen et al, 1992). The NDP protein plays a crucial role in the development of the retina, cochlea, and central nervous system by mediating the activation of the Wnt/b-catenin pathway and regulating cell division or differentiation (Warden et al, 2007; Xu et al, 2004). With the development of numerous related studies, various types of NDP defects have been identified including missense, nonsense, frameshift, or deletions.
Clinical exome sequencing is commonly defined as a targeted gene panel comprising most of the known human disease-causative genes in the Online Mendelian Inheritance in Man (OMIM) database. In addition to cost saving, clinical exome sequencing has deeper coverage and higher sequencing accuracy of targeted genes. Furthermore, the data sets of clinical exome sequencing that are functionally interpretable speed up the bioinformatics analyses and variant interpretation. In this study, we describe the clinical spectra of two unrelated Chinese families with ND and identify two NDP variants by using clinical exome sequencing.
Materials and Methods
Patients
Members of the two families with congenital blindness were referred to the Linyi People's Hospital. Familial pedigrees are shown in Figure 1. The ethics committee of the Linyi People's Hospital approved the research protocol, and written informed consent was obtained from every participant.
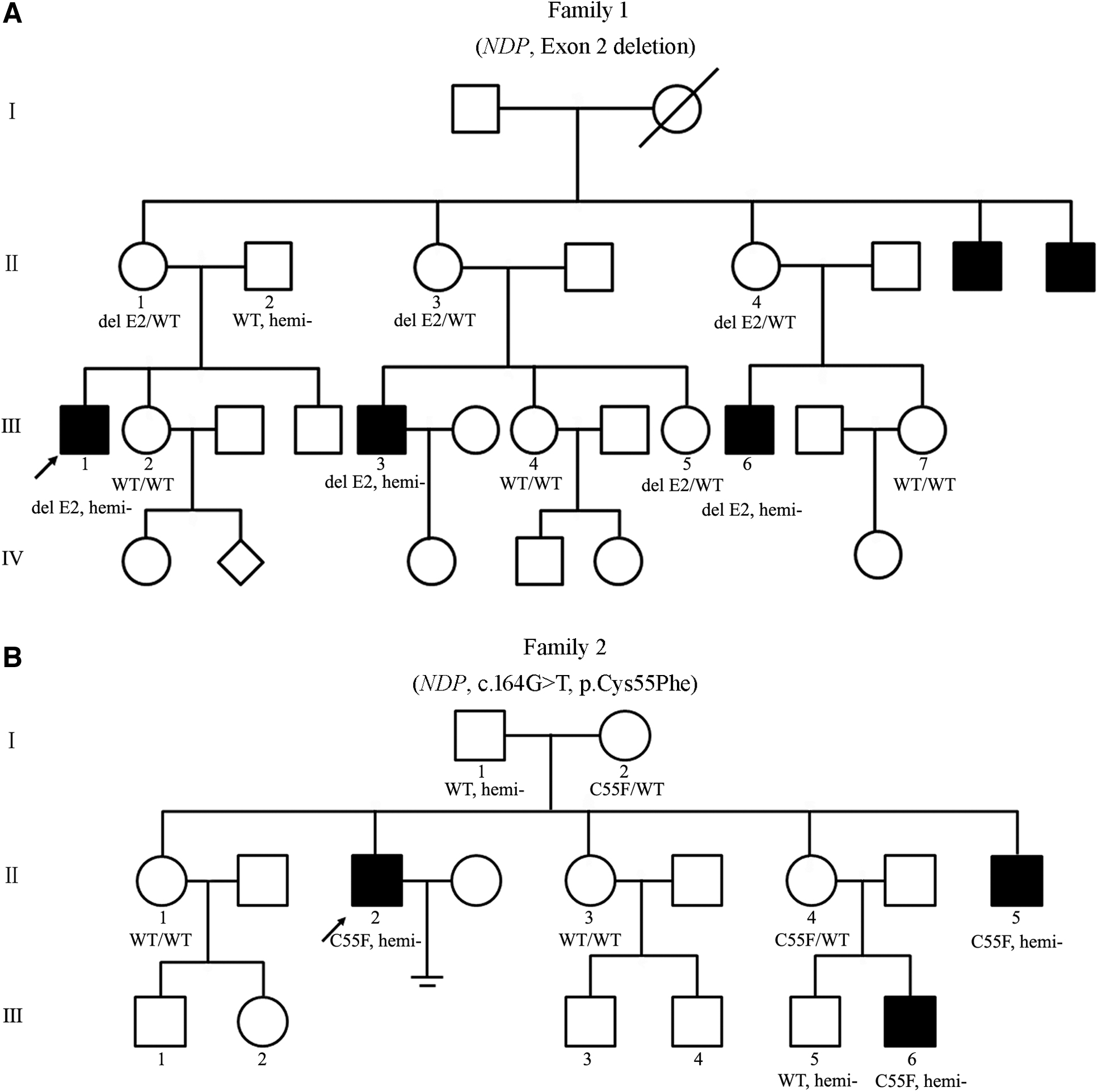
Pedigrees of the two families with ND.
Clinical exome sequencing
Genomic DNA samples of affected individuals, their family members, and healthy controls were extracted from peripheral blood using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according to manufacturer's instructions. Variant screening of ND patients was performed by clinical exome sequencing. The extracted DNAs were fragmented and then purified. DNA fragments were ligated with adaptors and captured by custom-designed NimbleGen SeqCap probes (Roche NimbleGen, Madison).
Enriched DNA samples were indexed and sequenced on a NextSeq500 sequencer (Illumina, San Diego). Sequencing reads were mapped to the reference human genome assembly (NCBI build 37/hg19). Nucleotide changes observed of aligned reads were called and reviewed by using NextGENe software (SoftGenetics, State College). Sequence variants were annotated using population and literature databases. Variants from the proband were processed together, and the source of each variant was annotated. After filtering out the synonymous and common single nucleotide polymorphism (SNPs) (minor allele frequency >0.1%), rare variants with high confidence were considered as the disease-causing candidate.
Variant annotation was further confirmed through literature and population databases, including 1000 Genomes, The single nucleotide polymorphism database, GnomAD, Clinvar, human gene mutation database, and OMIM. Genetic evaluation of pathogenicity of candidate variant genes was performed using multiple computational algorithms, including sorting intolerant from tolerant, Polyphen-2, and MutationTaster. The interpretation of variants was performed according to the American College of Medical Genetics (ACMG) standards and guidelines.
Sanger sequencing
Sanger sequencing was performed by polymerase chain reaction (PCR) and direct sequencing of the PCR product on 3500XL Genetic Analyzer (Applied Biosystems, Foster City). Sequences of the primers used for PCR were designed by Primer Premier 5.0. The primers for exon 2 deletion identification were forward (5′-CTGACGCTGACACCAAAG-3′) and reverse (5′-AGAGCCCTCCAAGAAGTA-3′). The primers for c.164G>T identification were forward (5′-GGAGGTGAAGCCATTTCCAATT-3′) and reverse (5′-CTTGCCTGTTTCTGAGGG-3′).
Results
Clinical features of the patients with ND
Patient III-1 in family 1 was a 35-year-old male. He was born at full term of normal weight and his mother denied history of drugs, alcohol, or illness during pregnancy. He was found to be blind in both eyes at early childhood and did not receive any eye examination until 35 years of age. Clinical examination showed no light sensation, enophthalmos, ocular shrinkage, corneal leukoma, and growth of massive neovascularizations, and other intraocular structures could not be observed (Fig. 2A, B).
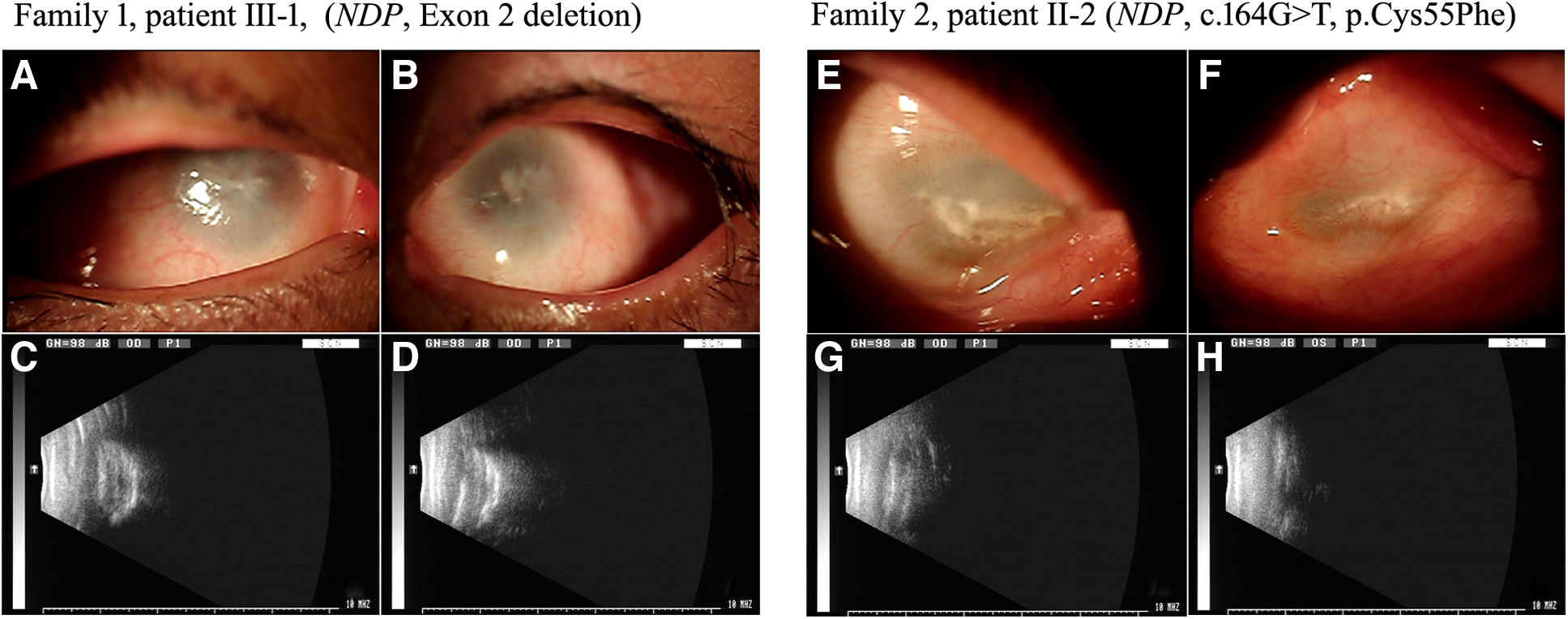
Photographs of the anterior segment and B-mode ultrasonography findings.
Moreover, this patient presented with mild sensorineural deafness. Brain magnetic resonance imaging was normal and mild cognitive impairment (22/30) was screened by using the minimal mental status examination. A family history revealed the presence of additional affected members all of whom were male (Fig. 1A). Combining clinical phenotypes and genetic pattern, ND was strongly suspected.
Patient II-2 in family 2 was a 31-year-old male. He was born at full term of normal weight after an uncomplicated pregnancy and his mother denied history of drugs, alcohol, or illness during pregnancy. He was found for not following moving light stimuli after birth by his parents and did not receive any eye examination until 31 years of age. Physical examination showed no light sensation, enophthalmos, severe ocular shrinkage, horizontal distribution of the central corneal porcelain white plaque, and growth of neovascularizations, and other intraocular structures could not be observed (Fig. 2E, F). Psychomotor development and auditive evaluation were normal in this subject. Because the affected members were all male, combining with retinal phenotypes, ND was also strongly suspected.
Variant analysis in NDP
To understand the possible genetic causes for the patients' retinal malformation, variant screening of DNA from patient III-1 (family 1) and patient II-2 (family 2) was performed by clinical exome sequencing. Finally, a fragment deletion containing the whole exon 2 and a missense variant (c.164G>T, p.Cys55Phe) of NDP (NM_000266.4) were identified, respectively (Fig. 3).
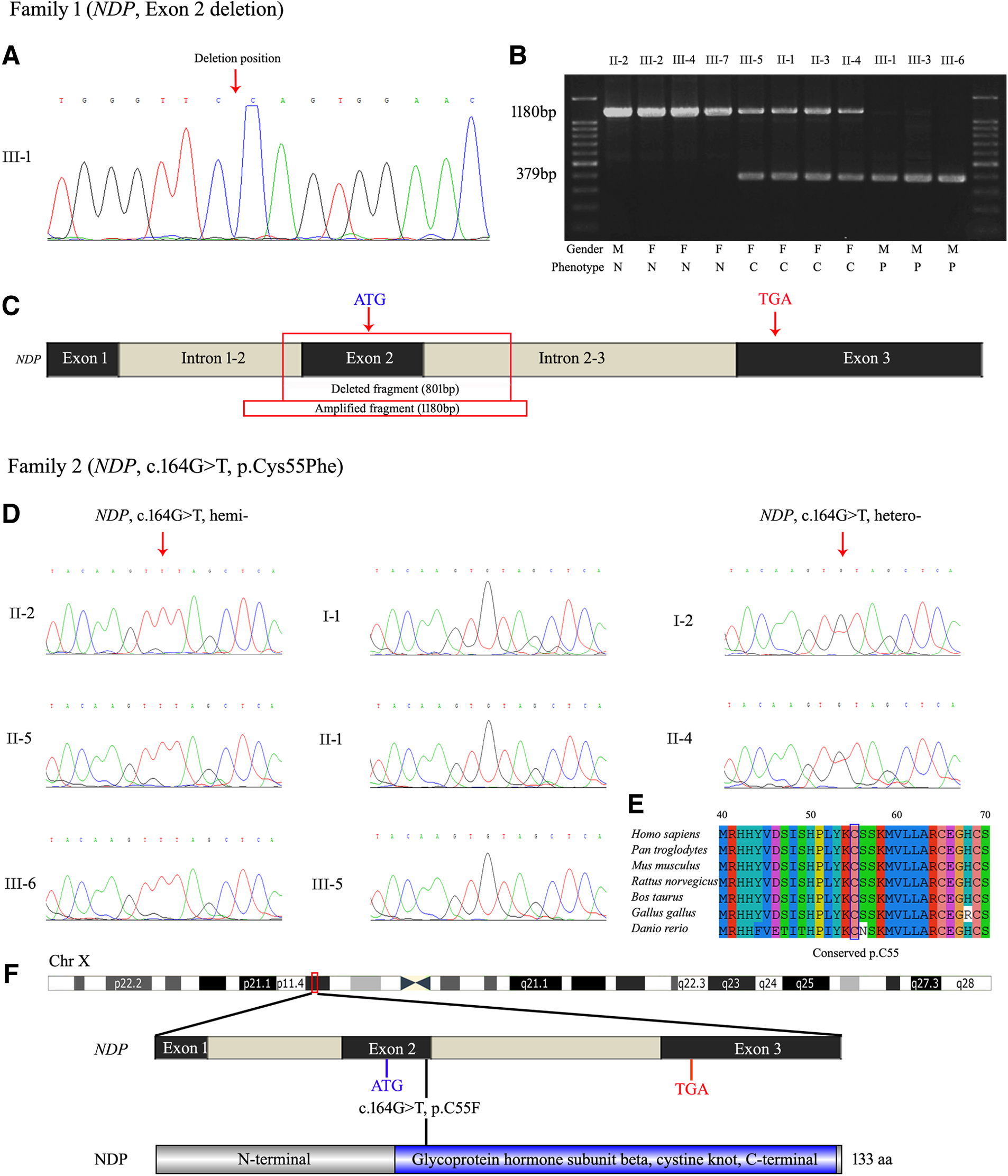
Variant and sequence analysis of NDP.
In family 1, we revealed the precise size of deleted fragment is 801 bp in patient III-1 by specific amplification and Sanger sequencing (Fig. 3A-C). Agarose gel electrophoresis demonstrated that the family members II-2, III-2, III-4, and III-7 are normal with one complete amplification band (1180 bp), the family members III-5, II-1, II-3, and II-4 are heterozygous female carriers with two amplification bands (1180 and 379 bp) and the family members III-1, III-3. and III-6 are hemizygous male patients with only one exon 2 deleted band (379 bp).
In family 2, Sanger sequencing revealed that the family members I-2, II-2, II-4, II-5, and III-6 carried the missense variant c.164G>T of NDP, whereas other unaffected members I-1, II-1, and III-5 lacked this variant (Fig. 3D). Thus, these two NDP variations showed the predicted segregation pattern for an X-linked pathogenic variant. Moreover, the missense variant c.164G>T of NDP was forecasted to be highly damaging to the physiological function of the NDP by PolyPhen-2 (0.998, deleterious) and PROVEAN (−2.695, deleterious). Alignment of NDP amino acid sequences in different species, including chimpanzee, mouse, rat, cow, chicken, and zebrafish, with the use of ClustalW2, showed high conservation of cysteine at position 55 (Fig. 3E, F).
Discussion
ND is an X-linked recessive retinal disorder involving severe ocular abnormalities. The ocular abnormalities include retinal dysplasia with early vascular proliferation, retinal detachment, cataract, atrophic irides, corneal opacification, and eventually ocular shrinkage. In addition to the ocular findings, several affected males may have varying degree of mental retardation, cognitive decline, and sensorineural deafness with age (Chen et al, 1992; Riveiro-Alvarez et al, 2005; Rodriguez-Munoz et al, 2018). In this study, we described two variations (c.164G>T, p.Cys55Phe and exon 2 deletion) in the NDP gene and the clinical spectra of two unrelated Chinese families with ND.
To date, the correlations between NDP gene variations and retinopathy have not been fully established. In consideration that NDP variants have been reported in ND, X-linked familial exudative vitreoretinopathy (XL-FEVR), persistent hyperplastic primary vitreous, retinopathy of prematurity (ROP), and Coats disease, it would be necessary to conduct detailed clinical examination before genetic analysis for a given case of retinopathy (Johnson et al, 1996; Shastry et al, 1997; Talks et al, 2001; Torrente et al, 1997).
Clinical exome sequencing is a subexome panel of capture-based NGS and commonly defined as a targeted gene panel comprising most of the known human disease-causative genes in the OMIM database. The clinical exome sequencing has become an ideal strategy for patient to identify phenotype-causing variants due to its high efficiency and accurateness in exome sequencing (Xu et al, 2017).
In this study, two probands from two unrelated Chinese families were diagnosed with ND based on their ocular manifestations, and pedigree investigation revealed an X-linked recessive pattern. Two variations identified in this study were c.164G>T, p.Cys55Phe and exon 2 deletion of NDP gene. The missense variant c.164G>T was reported in a previous study as a de novo variant in an uncomplicated family without genealogical cosegregation (Yang et al, 2012). In our case, the missense variant c.164G>T was identified from a relatively large family and showed the predicted segregation pattern for a pathogenic variant.
Moreover, we also predicted its pathogenicity and aligned its conservation in different species by in silico tools. Thus, the missense variant in exon 2 of NDP (NM_000266.4; c.164G>T, p.Cys55Phe) was classified as likely pathogenic (PS1 ± PM2 ± PP1 ± PP3) according to the ACMG standards and guidelines.
In addition, although the exon 2 deletion of NDP gene was qualitatively identified in several reported ND patients by fluorescence quantitative amplification or multiplex ligation-dependent probe amplification, we identified the precise size of deleted fragment by attempting multiple pairs of primers and showed the predicted segregation pattern for a pathogenic variant (Arai et al, 2014). Similarly, exon 2 deletion of NDP gene was classified as pathogenic (PVS1+ PS1+ PM4+ PP1+ PP4) according to the ACMG standards and guidelines.
Retinopathy is a genetically heterogeneous ocular malformation, different phenotypes such as XL-FEVR, ROP, or Coats disease may be due to the same variant of NDP gene (Arg121Trp) (Meindl et al, 1995; Shastry et al, 1997; Shastry et al, 1995). Besides the genetic heterogeneity occurring in ND, its clinical variability is also complex and may involve several molecular mechanisms apart from the NDP gene itself. In this study, two patients from two unrelated Chinese families were diagnosed with ND by eventually ocular shrinkage at a relatively high age, though this disease occurs early during embryogenesis and has prominent feature in newborn.
Moreover, both of these two families have no less than three patients. The reason behind this phenomenon is extreme lack of consciousness of birth defects prevention. In the perspective of a countryside family with specific genetic disease, a healthy member with certain educational background means the hope of problem solving is getting closer.
In conclusion, we described two variations (c.164G>T, p.Cys55Phe and exon 2 deletion) in the NDP gene and the clinical spectra of two unrelated Chinese families with ND. Our results broadened the mutational and phenotypic spectra of NDP and demonstrated the feasibility of clinical exome sequencing in clinical application of molecular diagnosis.
Footnotes
Authors' Contributions
X.Z. and Q.L. designed research. X.Z., C.G., L.L., and L.J. performed main experiments. Y.W. and F.C. did the bioinformatical analysis. X.Z., F.C., and Q.L. wrote the article. All authors critically analyzed, discussed, and interpreted data, and edited the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The study was supported by the Shandong Provincial Natural Science Foundation, China (Grant Nos. ZR2020QH189, ZR2020QH047, and ZR2021QH268).