
Abstract
Background:
Deleterious mutations in the human gene phenylalanine hydroxylase (PAH) encoding the phenylalanine hydroxylase enzyme give rise to classic phenylketonuria and hyperphenylalaninemia. Our study was designed to characterize the spectrum of variants in the PAH gene in Saudi patients.
Materials and Methods:
We screened a cohort of 72 Saudi patients with clinical and biochemical diagnoses of hyperphenylalaninemia at the largest tertiary care center in Saudi Arabia; the King Faisal Specialist Hospital and Research Center (KFSH&RC), Riyadh. All patient's charts were reviewed under an approved study by Institutional Review Board.
Results:
Twenty-one different PAH variants were identified among the 144 PAH alleles assessed by targeted gene sequencing. Within the studied cohort, 60 of 72 patients had homozygous mutations with the the remaining 12 being compound heterozygotes. The most prevalent of the disease alleles identified in this study was the p.(Arg252Trp) mutation, which accounted for 38 of 144 alleles (26.4%). With the high incidence of genetic disorders in the population, religiously permissible preventive reproductive measures are a priority in our practice. Prenatal diagnoses carried out on four fetuses revealed two that were homozygous for PAH pathogenic variants. In addition, pre-implantation genetic diagnoses were initiated for 19 families. Eight of these families completed more than one full cycle of treatment, from which one healthy newborn was delivered.
Conclusions:
This study describes the spectrum of PAH variants in the Saudi population and highlights the molecular heterogeneity underlying phenylketonuria and hyperphenylalaninemia. These results add to the existing knowledge about PAH variants in Middle Eastern Countries. These results can be further translated to provide: informed counseling; cascade carrier testing in extended family members; and pre-marital screening.
Introduction
Hyperphenylalaninemia (HPA)
Patients with PAH deficiency can be classified into two groups: (1) not requiring treatment and (2) requiring diet, BH4, or both. The treatment is not recommended if the blood Phe level in untreated patients <300 μM. All untreated patients with blood Phe level >360 μM should be treated. Patients with untreated Phe levels between 360 and 600 μM should be treated until 12 years of age. Patients with untreated Phe levels >600 μM should be treated for life (Blau et al., 2011; Van Spronsen et al., 2017; Van Wegberg et al., 2017).
Folling first described (PKU; OMIM 261600) in 1934 when he successfully detected phenylketone bodies in the urine of affected individuals (Christ, 2003). Bickel later reported the effective use of a low-Phe diet in 1953 (Bickel et al., 1953). In the 1960s, a major contribution to preventing intellectual disability was achieved through Guthrie's invention of a simple test to detect HPA (Guthrie and Susi, 1963).
Subsequently, PKU was the first condition to be added to the list of newborn screening (NBS) programs. However, after more than 50 years of establishing NBS for PKU, it is not available in many parts of the world. Therefore, policy makers in those countries should implement it to prevent severe neurocognitive and neuromotor impairment in affected neonates (Berry et al., 2013; Van Spronsen et al., 2017).
Based on data collected from the National Newborn Screening Program in Saudi Arabia, the estimated overall incidence of inborn errors of metabolism is 1:1143 (Alfadhel et al., 2017). This suggested incidence is considered high compared with worldwide figures due to the high rate of consanguinity in Saudi Arabia (Al-Owain et al., 2012). Consanguineous unions are favored in Arab communities and account for 20-50% of marriages.
The occurrence of PKU varies among ethnic groups and geographic regions worldwide. The highest prevalence was reported in European and certain Middle Eastern populations. Italy (1:4000) and Ireland (1:4545) had higher prevalence than Iran and Jordan (both 1:5000). Northern Europe showed the lowest PKU rates in Europe, for example, the prevalence is 1:112,000 live births in Finland and 1:13,434 in Denmark (Hillert et al., 2020).
Few countries in the region, such as Turkey, Saudi Arabia, United Arab Emirates, and Qatar, have established a comprehensive NBS program, which includes PKU, among other conditions (El-Metwally et al., 2018). The highest rate of PKU was reported in Turkey, the Fars province of Iran, and the Russian republic of Karachay-Cherkessia, which is estimated to be 1:6667, 1:4698, and 1:850 newborns, respectively (Gundorova et al., 2018; Hillert et al., 2020; Senemar et al., 2011).
In Saudi Arabia, HPA in newborns had an approximate incidence of 1:14,245 (Alfadhel et al., 2017). Other Arab countries had lower PKU prevalence such as Iraq (1:14,286) and United Arab Emirates (1:14,493) (Hillert et al., 2020). There is a lack of published data on the prevalence of PKU in Arab countries, such as Algeria, Syria, Libya, Sudan, and Yemen, most likely due to the absence or limitation of screening programs (Therrell et al., 2015). The low prevalence of PKU reported for South America ranges from ∼1:25,000 to 50,000 live births (Borrajo, 2007; Hillert et al., 2020).
In many populations, most cases of PKU are detected through NBS programs, and dietary treatment is started promptly (Vardey et al., 2020). As a result, severe signs and symptoms of the disease are not frequently seen. Without dietary restriction, patients present with variable intellectual disability, microcephaly, autism, seizures, and behavioral and psychiatric symptoms (Blau et al., 2010).
The human PAH gene is located on the long arm of chromosome 12, comprising 13 exons, and spans 90 kb, encoding a protein of 452 amino acids. It is the only known gene causing PAH deficiency. To date, around 1723 PAH variants (PAHvdb database; www.biopku.org/home/pah.asp; as of March 7, 2023) are known to be associated with PAH deficiency.
Population-based founder variants have been reported, including a deletion spanning the third exon of the PAH gene among the Yemenite Jewish population (Bercovich et al., 2008). Moreover, deletion of exon 3 accounts for 9% of the identified PKU variants in Cypriot patients, detected through neonatal screening (Georgiou et al., 2012).
This study's objectives were to characterize the spectrum of variants in the PAH gene in Saudi patients and to expand the knowledge about PAH gene variants in the Saudi population with a confirmed diagnosis based on biochemical, clinical, and molecular findings.
Materials and Methods
Patients and PKU diagnosis
This retrospective study included a cohort of 72 unrelated patients diagnosed with HPA using molecular genetic testing at the largest tertiary care center in Saudi Arabia, King Faisal Specialist Hospital and Research Centre (KFSH&RC), Riyadh. The PAH gene variant analysis was carried out previously at routine patient visits to genetics clinics during childhood. Targeted prenatal molecular diagnosis was performed for four expectant mothers.
In addition, a preimplantation genetic diagnosis (PGD) referral was initiated for 19 mothers of patients from this study. Patients' charts were reviewed as part of an Institutional Review Board (IRB)-approved research project (RAC No. 2171027).
Variant detection and prenatal sampling
Genomic DNA was extracted using the PUREGENE DNA Extraction Kit according to the manufacturer's instructions (Gentra Systems, Minneapolis, MN). Gene-specific intronic primers were designed to flank each of the 13 coding exons of the PAH gene (GenBank accession No.: NM_000277.1) using the Primer3, v.0.4.0, program. Amplification by PCR was performed in a total volume of 25 μL, containing 20 ng DNA and recommended amounts of Deoxynucleoside triphosphates (dNTPs), primers (Metabion, Martinsried, Germany), and HotStar Taq DNA polymerase (Qiagen, Germantown, MD).
PAH primer sequences and PCR conditions are available on request. Purified PCR products were then sequenced with a BigDye™, v3.1, Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA), and products were read using an ABI PRISM 3730 DNA analyzer (Applied Biosystems). Sequencing chromatograms were compared with the published gDNA sequence using the SeqMan 6.1 module of the Lasergene software package (DNAStar, Inc., WI, USA). Deletion/duplication analysis and SNP array techniques were performed at Baylor College of Medicine using array-based comparative genomic hybridization (aCGH).
Chorionic villus sampling (CVS) between 10 and 12 weeks was performed on four expectant mothers who previously had an affected child with biallelic pathogenic PAH variants indentified by this study. Fetal genomic DNA was extracted and targeted variant analysis was performed for the specific variant using PCR and direct Sanger sequencing.
Results
A total of 72 Saudi patients (29 males and 43 females) were included in this study. The age of our cohort ranged between 7 and 35 years. The diagnosis was made based on the biochemical finding of high blood Phe level and molecular confirmation of PAH variants (Table 1).
Phenylalanine Hydroxylase Gene (PAH) Variants in 72 Saudi Patients with Hyperphenylalaninemia
These 12/72 patients with PKU harbor compound heterozygous PAH variants. The second variant in two patients (cases 24 and 29) was not identified. Rest of the 60/72 patients had variants in the homozygous state.
NA, not applicable; PAH, phenylalanine hydroxylase; Phe, phenylalanine; PKU, phenylketonuria.
The molecular basis of HPA was established for our cohort by direct sequencing of the entire coding region and intron-exon boundaries of PAH. In total, we identified 21 different variants across the PAH gene in our cohort (Fig. 1; Table 1). All variants were previously reported. The 4.59 kb deletion was detected by aCGH and included part of exon 3 with breakpoints in intron 2 and in exon 3 corresponding to a minimum deletion boundary through aCGH of arr[GRCh37]12q23.2(103,288,901-103,293,251)x0.
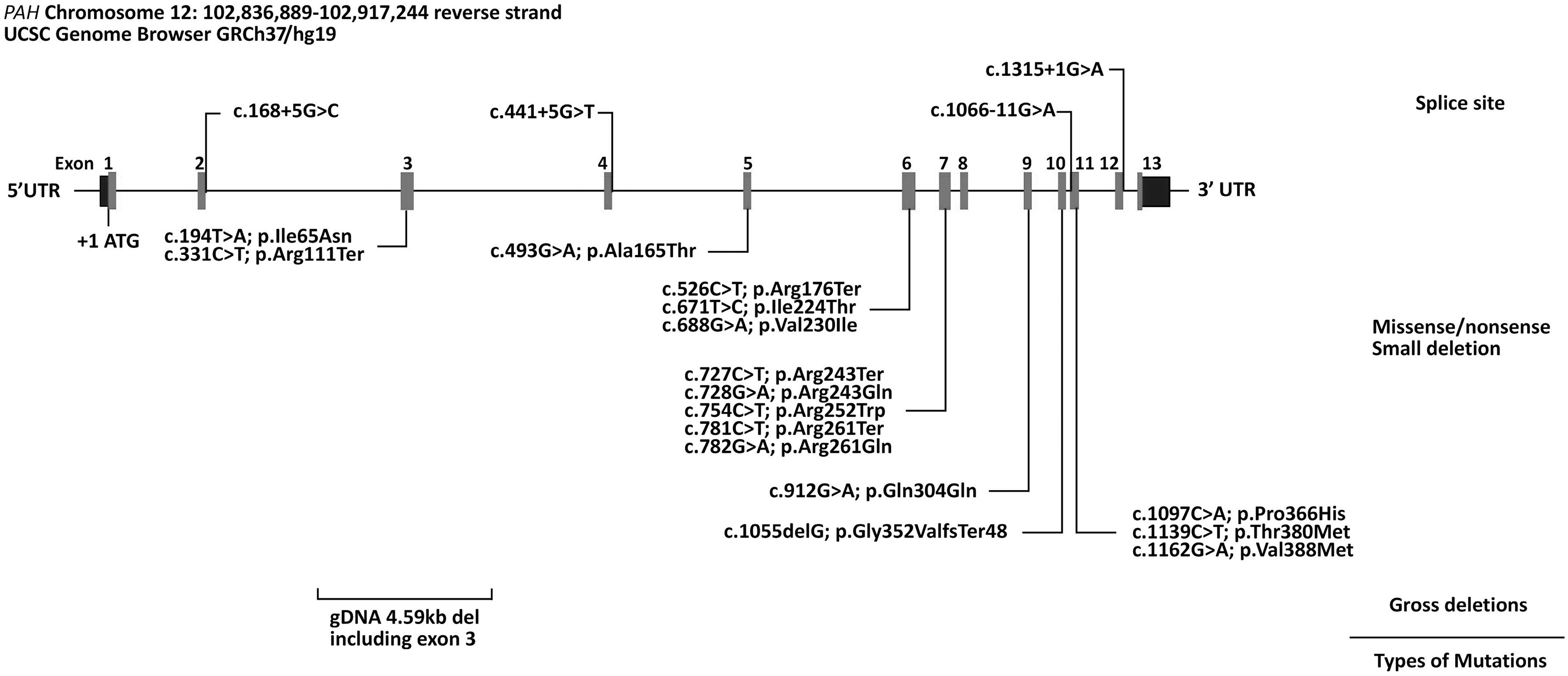
Schematic representation of variants in the PAH gene identified in our cohort of 72 Saudi patients with PAH deficiency. The reference accession number for the PAH sequence is NM_000277.1 and for the encoded protein is NP_000268. Exons of the PAH gene are indicated by boxes and introns by interconnecting lines. Splice site mutations are grouped above, while missense/nonsense variants and small and gross deletions are grouped below the graphic illustration of the gene.
PCR and sequence analysis across the 4.59 kb deletion using custom-designed primers confirmed deletion of part of exon 3, including the acceptor site and upstream intronic region in conjunction with insertion of 66 nucleotides in these individuals, PAH: c.169-4591_171delins66. The individual genotyping profiles of our 72 Saudi patients with HPA are summarized in Table 1.
Genotypes of 60 of 72 (83.3%) patients were in a homozygous state and 12 of 72 (16.7%) were compound heterozygous. Homoallelic variants were observed in most of our patients, reflecting the high rate of consanguinity in the Saudi population. The majority of the total types of variants identified were missense (11 of 21), nonsense (4 of 21), and splicing (4 of 21), and there was (one of both) a small and a large deletion. The p.(Arg252Trp) was the most prevalent variant in our cohort with an allele frequency of 26.4% (38 of 144 alleles).
In addition, three other variants, p.(Arg261Ter), p.(Val388Met), and c.168 + 5G>C, were found at relatively high frequencies (10.4%, 9.7%, and 9%, respectively). The c.1066-11G>A, p.(Ile224Thr), 4.59 kb deletion, p.(Arg243Ter), p.(Arg261Gln), and p.(Arg111Ter) variants were less frequent. All other variants had frequencies of 2% or less (Table 2).
Relative Allele Frequencies of Phenylalanine Hydroxylase Gene Mutations in 72 Independent Saudi Phenylketonuria Patients
Sequencing of the PAH gene failed to identify the second variant in two patients (cases 24 and 29) clinically known to have PKU (Table 1). Deletion/duplication analysis was negative for Patient 24, but not performed for Patient 29 as patient contact was lost during follow-up.
Results of prenatal genetic testing on CVS in four expectant mothers who previously had an affected child, with biallelic pathogenic variants identified by this study, are shown in Table 3. Two fetal samples were determined to be either wild-type normal or heterozygous carriers for the target variant, and the remaining two of four fetuses were homozygous for the pathogenic p.(Arg261Ter) and c.1066-11G>A variants in the PAH gene. In all cases, the pregnancies continued successfully.
Results of Prenatal Genetic Testing of Targeted Variants from Expectant Mothers with Affected Phenylketonuria Children Identified by This Study
CVS, chorionic villus sampling.
In addition, 19 mothers of patients from this study were referred for PGD at KFSH&RC. Eleven of these were no-shows at the clinic. The remaining eight mothers were documented to have completed at least one round of treatment, from which one healthy newborn was delivered.
Discussion
This study describes the spectrum of variants in the PAH gene among 72 Saudi HPA patients diagnosed based on their abnormal tandem mass spectrometry (MSMS) profile and molecular confirmation of pathogenic variants in the PAH gene. Our study highlights the heterogeneity of HPA variants in this population.
All patients in our cohort had a blood Phe level above 360 μM and therefore required treatment. The individual genotyping profiles of our 72 Saudi patients with HPA are summarized in Table 1.
Among the molecular profiles of our patients, the p.(Arg252Trp) variant was predominantly identified in this cohort, followed by p.(Arg261Ter), p.(Val388Met), and c.168 + 5G>C variants. p.(Arg252Trp) is located at the catalytic domain of the PAH gene and was found in 20 of 72 patients (27.8%). The p.(Arg261Ter) variant was seen in 9 of 72 (12.5%) patients and the splice site variant, c.168 + 5G>C, was seen in 8 of 72 (11.1%) patients in our cohort. The remaining variants were less common.
In contrast to our study, 124 Iranian patients with classic PKU were included in a study to identify the molecular basis of PKU. The predominant variant in that study was IVS10-11G>C, with a frequency of 24.6%. This variant was prevalent in the Mediterranean population, which shares the same geographical and historical link (Zare-Karizi et al., 2011).
Similarly, a study included 90 unrelated Egyptian patients tested for six common Mediterranean variants, and the authors found that the IVS10-11G>C variant was common with a frequency of 17%, while the p.(Arg252Trp) variant was less common, representing a frequency of 1.6% (Effat et al., 2008).
The c.168 + 5G>C variant seen in our cohort has been described earlier in patients from Lebanon (Karam et al., 2013) and other Mediterranean countries. However, this variant was seen at a lower frequency in Iranian patients (Zare-Karizi et al., 2011) and in patients from the Kemerovskaya region and Sakha Republic of Russian Federation (Baturina et al., 2012). Sequencing of the PAH gene failed to identify the second variant in two patients (cases 24 and 29). This may be due to the possibility that it is located deep in the intronic or 3′ UTR regions.
Kuvan (sapropterin dihydrochloride) is a form of BH4, the cofactor of the enzyme PAH, which helps the enzyme to break down Phe. It is used as a treatment modality for PKU, in addition to a low-protein diet and special formula. All patients with PKU in our cohort are on a low-protein diet. Kuvan responsiveness was assessed in some patients in our cohort; however, we needed more regular follow-ups to generate reliable data.
Therefore, genotype-based prediction of BH4 responsiveness was established based on the BIOPKU database (www.biopku.org/biopku). The majority of the genotypes in our cohort were unresponsive. Only four genotypes are responsive to Kuvan, according to the BIOPKU database, including p.(Val230Ile), p.(Arg261Gln), p.(Gln304Gln), and p.(Val388Met).
With the high incidence of genetic disorders, religiously permissible, preventive reproductive measures are a priority in our practice and widely requested by Saudi couples with a history of genetic diseases (AbdulAzeez et al., 2019; Alsulaiman et al., 2010). Identification of causative variants is essential to offer timely and accurate prenatal diagnosis (PND) and PGD. Among the four prenatal cases described in Table 3, two fetuses were found to be homozygous for known PAH pathogenic variants. This allowed for immediate implementation of dietary restrictions after birth.
Eight mothers from this study underwent PGD. Three mothers completed three in vitro fertilization (IVF) cycles, four mothers completed one IVF cycle, and the remaining mother completed two IVF cycles. To date, only one living healthy child was born post-PGD. In the remaining cases, there was either no suitable embryo for transfer or the pregnancy test was negative. PKU is a chronic condition treatable through lifelong dietary management. Heterozygous parents have a 25% risk of recurrence in a subsequent pregnancy.
During a genetic counseling session, genetic counselors offer preventative measures for the couple. Some couples accept the recurrence of the disease because they have an affected child on a restricted diet that yielded good outcomes. Other couples opt for PND or PGD based on the regulation of termination of pregnancy in our hospital or the policy for acceptance at the PGD unit. In Saudi Arabia, PND is an option for couples to prevent the recurrence of PKU. Similarly, PND for PKU was performed and reported in several studies.
In one study, the PND showed that 3 of 24 fetuses were affected, 8 were wild type, and 13 were heterozygous carriers (Yan et al., 2019). Invasive and noninvasive PND results for pregnant women of PKU families were concordant in several studies, with no false-positive or false-negative results (Peng et al., 2021; Yan et al., 2019). PGD is an alternative preventative reproductive approach for parents who wish to eliminate the chance of having an affected child with PKU.
PGD is a widely used method for prevention in Saudi Arabia. However, long waiting times and failed treatment cycles result in parents conceiving normally without intervention. The Human Fertilisation and Embryology Authority licensed PGD for PKU in the United Kingdom. Likewise, PGD is widely accepted for lethal conditions, but there is an ongoing debate about the use of PGD to test for chronic disorders such as PKU, where early diagnosis and effective management are available.
Approval for the use of PGD for PKU in the United Kingdom was obtained after several reviews concerning the physical and psychosocial outcomes of patients with PKU and their families (Lavery et al., 2013). For example, a 31-year-old woman with a child affected with PKU was offered PND by a health care facility. She declined PND and opted for PGD. She went through PGD cycles, and an unaffected blastocyst was transferred on day 5 to her uterus, resulting in a clinical pregnancy. The outcome was a healthy boy. The remaining unaffected and carrier blastocysts were cryopreserved (Lavery et al., 2013).
Conclusions
In our study, we detected 21 variants in the PAH gene, which have been previously reported. The spectrum of these variants in Saudi patients confirms the heterogeneity of our mixed population. Our results can be used as a source to streamline the plan for future treatment and family counseling. In addition, identification of pathogenic variants causing HPA will be of tremendous use for future carrier testing, prenatal testing, premarital screening, and PGD in Saudi Arabia and neighboring countries.
Footnotes
Acknowledgments
The authors thank the sequencing core facility for their technical help. The authors also thank the families for their participation in the study.
Authors' Contributions
A.B. and M.A. were primarily responsible for clinical support, clinical evaluation, and sample collection. A.B., F.I., K.R., and S.A. were responsible for molecular genetic studies, data interpretation, and drafting the manuscript. A.B., F.I., K.R., and M.A. participated in the final editing of the manuscript. All authors have read and approved the final manuscript.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was funded by King Faisal Specialist Hospital and Research Centre (KFSH&RC-(RAC # 2171027).