
Abstract
Objective:
Polydactyly is characterized by multiple distinct heterogeneous phenotypes, the etiologies of which involve several genes. This study aimed to explore the genetic defects and further clarify the molecular mechanism of polydactyly in several Chinese families.
Methods:
Three families with diverse phenotypes of non-syndromic polydactyly were analyzed: two were cases of familial disease, whereas one was sporadic. PCR and Sanger sequencing were used to screen for pathogenic mutations in two known disease-associated genes, GLI3 and HOXD13, while bioinformatic analyses predicted the pathogenicity of the identified variants. Reverse transcription PCR was used to analyze the splicing effect of an intronic variant.
Results:
Two novel heterozygous frameshift mutations (c.4478delG/p.S1493Tfs*18; c.846_c.847insC/p.R283Qfs*21) were identified in the GLI3 gene from two of the pedigrees. Both c.4478delG and c.846_c.847insC were later confirmed in affected and unaffected members and normal controls, to truncate and disrupt the integrity of the GLI3 protein, reduce its level of expression, and disrupt its biological function through nonsense-mediated mRNA decay (NMD). In addition, a deep intron mutation (c.125-47 C>A) was detected in the GLI3 gene from the sporadic case, however, both bioinformatics analysis (HSF, splice AI, and CBS) and RT-PCR indicated that the variant c.125-47 C>A had minimal if any impact on splicing of the GLI3 gene.
Conclusion:
Two newly identified heterozygous frameshift mutations in the GLI3 gene were detected in two families with non-syndromic polydactyly, further extending the mutational spectrum of the GLI3 gene in non-syndromic polydactyly. Moreover, our study further expanded the phenotypic spectrum of non-syndromic polydactyly.
Introduction
Polydactyly is one of the most common limb anomalies, with a population incidence of ∼1.6-10.7 in 1000 live births (Malik, 2014; Umair et al., 2018). Duplication of one or several digits and syndactyly of multiple digits are the two main features in individuals with polydactyly. This deformity usually appears in two forms: non-syndromic and syndromic. Syndromic polydactyly is characterized by malformations in digits and other organs or systems (Biesecker, 2002). According to the involved digits, non-syndromic polydactyly is classified into three categories: preaxial polydactyly (PPD), postaxial polydactyly (PAP), and central polydactyly.
In PPD and PAP, malformed fingers or toes are located on the radial and ulnar side, respectively (Fujioka et al., 2005). PAP is subdivided into postaxial type A (PAPA) in which the extra digit is well developed, and postaxial type B (PAPB) in which the extra digit is hypoplastic rudimentary or appears as a skin tag or a small protuberance (Umair et al., 2019).
In most non-syndromic polydactyly families, transmission occurs in an autosomal dominant manner; however, a small number of families with non-syndromic polydactyly are characterized by autosomal recessive inheritance (Mollica et al., 1978; Radhakrishna et al., 1997). To date, six genes or genomic regions associated with non-syndromic polydactyly have been identified in humans, namely, GLI3, SHH/ZRS, MIPOL1, ZNF141, PITX1, and IQCE, with GLI3 and SHH/ZRS being the most commonly reported genes (Deng et al., 2015; Umair et al., 2017).
However, other genes, including HOXD13, FBLN1, LMBR1, and SHH, have also been found to be direct causative agents of non-syndromic polydactyly (Debeer et al., 2002; Niedermaier et al., 2005; Park et al., 2008; Zhao et al., 2007). Genetic variants in any of these genes might result in limb malformations. Mutations in the HOXD13 gene have been closely related to limb deformities both in vivo and in vitro.
Expansion or contraction of the polyalanine tract in exon 1 of the HOXD13 gene usually causes typical synpolydactyly (SPD), whereas some deletions, missense, or splice mutations in other coding regions lead to atypical SPD (Goodman et al., 1998; Kan et al., 2003; Kjaer et al., 2002). The phenotypes of non-syndromic polydactyly are distinctly interfamilial and intrafamilial heterogeneous, thus complicating the analysis of the relationship between genotype and phenotype and the delineation of the underlying molecular mechanism.
Genotype and phenotype correlation studies have indicated that germline mutations in the GLI3 gene are associated with syndromic and non-syndromic polydactyly, including Pallister-Hall syndrome (PHS), Greig cephalopolysyndactyly syndrome (GCPS), and isolated polydactyly (Debeer et al., 2007; Johnston et al., 2005; Kalff-Suske et al., 1999; Kang et al., 1997; Radhakrishna et al., 1999). Moreover, mutations at different sites of GLI3 gene have been shown to potentially lead to different types of symptoms.
Most patients with GCPS carry mutations in the 5′ and 3′ ends of GLI3 gene, whereas patients with PHS are more prone to have mutations in the central part of the gene (Démurger et al., 2015; Jamsheer et al., 2012). Isolated polydactyly caused by GLI3 gene mutations is sporadically reported without clear and preferred domains or locations of the GLI3 gene (Al-Qattan, 2012; Cheng et al., 2011; Fujioka et al., 2005). As the molecular genetic mechanism remains ambiguous, more studies are needed.
In this study, we investigated three Chinese families or cases with hereditary non-syndromic polydactyly and ultimately detected two novel frameshift mutations in the GLI3 gene in two of these families.
Materials and Methods
Subjects and samples
Families and sporadic cases with isolated or non-syndromic polydactyly were recruited from Fujian Province with Han Chinese origin. Proband 1 and 2 were familial and transmitted in an autosomal dominant inheritance mode, whereas proband 3 was a sporadic case. In addition, normal controls were recruited from the medical examination center.
All participants were subjected to comprehensive assessment by clinical and radiological examinations and pedigree investigation. All participants signed an informed consent, and 2-3 mL of peripheral blood was drawn from each individual and temporarily stored at 4℃. The study was approved by the Ethics Committee of Fuzhou Second Hospital (Approval No. 2022001). All procedures complied with the principles listed in the Declaration of Helsinki.
Polymerase chain reaction and Sanger sequencing of GLI3 and HOXD13 genes
Considering that mutations in the HOXD13 gene were also common causative agents in patients with polydactyly, we thus analyzed both the coding regions of the GLI3 and HOXD13 genes. Samples of peripheral blood from proband 1, proband 2 and the parents, proband 3 and normal controls were processed and total DNA was extracted using the SE Blood DNA Kit (Omega BioTek, Norcross, Georgia, USA) according to the manufacturer's instructions.
Coding regions of the two genes of three probands were first amplified. The mutated sites of the parents and normal controls were tested and verified after the pathogenic mutations were identified in the probands. The polymerase chain reaction (PCR) amplification system was performed, and the cycling conditions for the amplification of the GLI3 and HOXD13 genes (the fragments of exon 1 and exon 2) were as follows: initial denaturation at 94°C for 2 min, followed by 30 cycles of denaturation at 94°C for 30 s, annealing for 30 s, extension at 72°C for 30 s; and a last extension cycle at 72°C for 2 min.
The fragments with triplet repeat in exon 1 of the HOXD13 gene were also amplified with 2 × PrimeSTAR GC buffer (Mg2+plus) and PrimeSTAR HS DNA polymerase according to the manufacturer's instructions (Takara Bio, Inc.). PCR conditions were as follows: initial denaturation at 95℃ for 3 min, followed by 35 cycles of denaturation at 95℃ for 30 s, annealing at 60℃ for 30 s, extension at 72℃ for 1 min, and final extension at 72℃ for 10 min.
All PCR products were separated on a 1.5% agarose gel and then viewed using an automatic gel imaging system (GIS 3500) (Tanon Biotechnology, Shanghai, China). Purified products were sequenced on an ABI 3730 XL genetic analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
In silico analysis and prediction
According to the standard guidelines and recommendations of the American College of Medical Genetics and Genomics (ACMG), candidate variants were classified into five types: pathogenic, likely pathogenic, uncertain significance, likely benign, and benign (Richards et al., 2015). All candidate variants in disease-associated genes were picked out and assessed based on the ACMG guidelines. In addition, three bioinformatic databases, HSF (Version 3.1) (http://www.umd.be/HSF3/), spliceAI (https://pypi.org/project/spliceai/), and CBS (NetGene2 Server) (http://www.cbs.dtu.dk/services/NetGene2/), were used to predict the probable pathogenicity of splicing and intronic variants.
Reverse transcription PCR analysis for intronic variant
RNA was drawn from proband 3 with deep intronic variant and normal control using RNAprep Pure Hi-Blood Kit (TIAN GEN, Beijing, China), according to the manufacturer's instructions. RNA was reversely transcribed into cDNA (Takara Bio, Inc.), which was used as the template for the following RT-PCR. The primers for reverse transcription-PCR (RT-PCR) were designed in exon 2 and exon 4 of GLI3 gene mRNA (NM_000168) (F: GCTCCACGACCACTGAAAAG; R: GCAATGGAGGAATCGGAGAT), and the fragment size was 450 bp. Purified products were viewed and sequenced as described earlier.
PCR and Sanger sequencing of the GLI1 and IQCE genes for proband 3
It is known that mutations in GLI1 and IQCE genes are also known to cause PAP; coding regions of these two genes were also analyzed by PCR and Sanger sequencing, and the conditions were stated earlier.
Results
Physical, imaging examinations, and pedigree investigation
We subjected all participants to careful physical and imaging examinations, and we performed pedigree investigation. The proband in pedigree 1, 3 members (the proband, father, and mother) in pedigree 2, and the proband in pedigree 3 were included in this study.
We confirmed the digital phenotypes of the probands by both physical and imaging examinations; the other members were diagnosed based on physical examinations and pedigree investigation (Table 1; Fig. 1). All three pedigrees exhibited non-syndromic polydactyly: pedigree 1 was PAP, in which the mother had typical symptoms, whereas pedigree 2 exhibited a preaxial-PAP syndactyly complex that was mainly found in the father (Table 1).
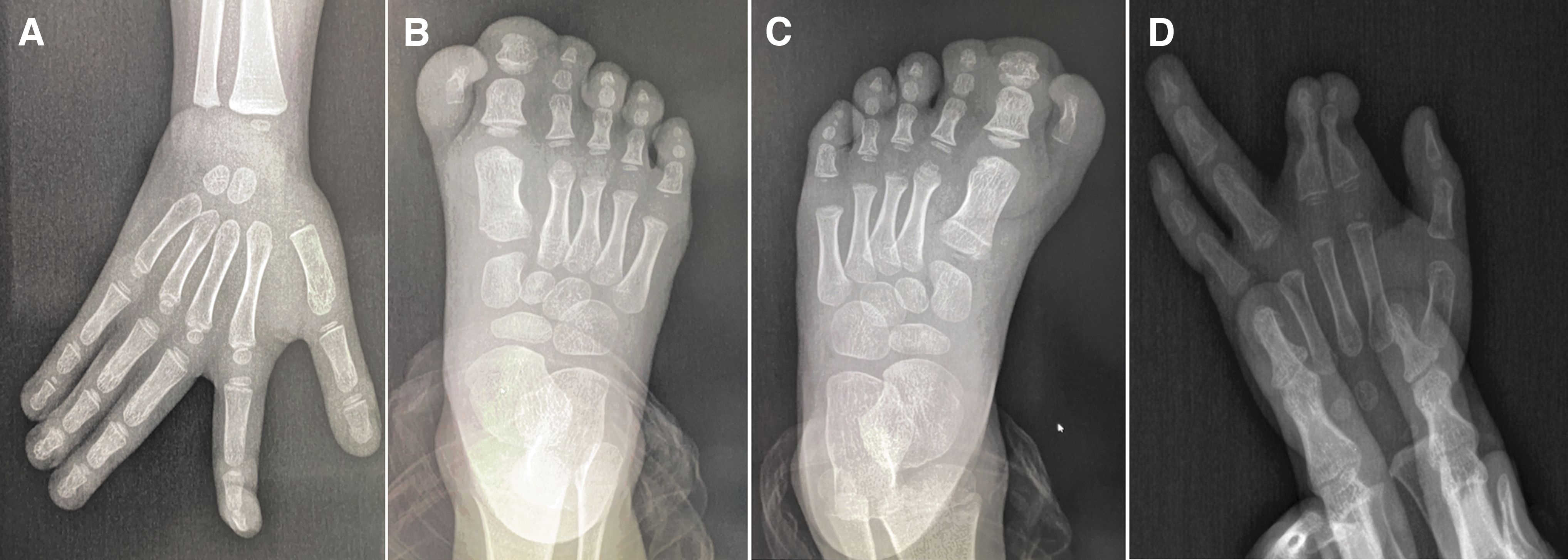
Imaging examinations of three probands with non-syndromic polydactyly.
Clinical Features of the Pedigrees with Polydactyly
PCR and Sanger sequencing of coding regions of GLI3 and HOXD13 genes
Considering the three probands were presented with non-syndromic polydactyly, thus we amplified GLI3, the first candidate gene. We separately identified two pathogenic mutations and one intronic mutation with uncertain pathogenicity in the GLI3 gene of these three probands (Fig. 2; Table 2). In particular, we detected the c.4478delG/p.S1493Tfs*18 in proband 1 and c.846_c.847insC/p.R283Qfs*21 in proband 2, which were identified as two novel frameshift mutations that produce premature stop codons and ultimately truncate the protein. In addition, we identified the c.846_c.847insC/p.R283Qfs*21 in the father of proband 2, whereas the mother carried wild-type alleles in this site, conforming to the phenotypes of pedigree 2 and autosomal dominant inheritance mode.
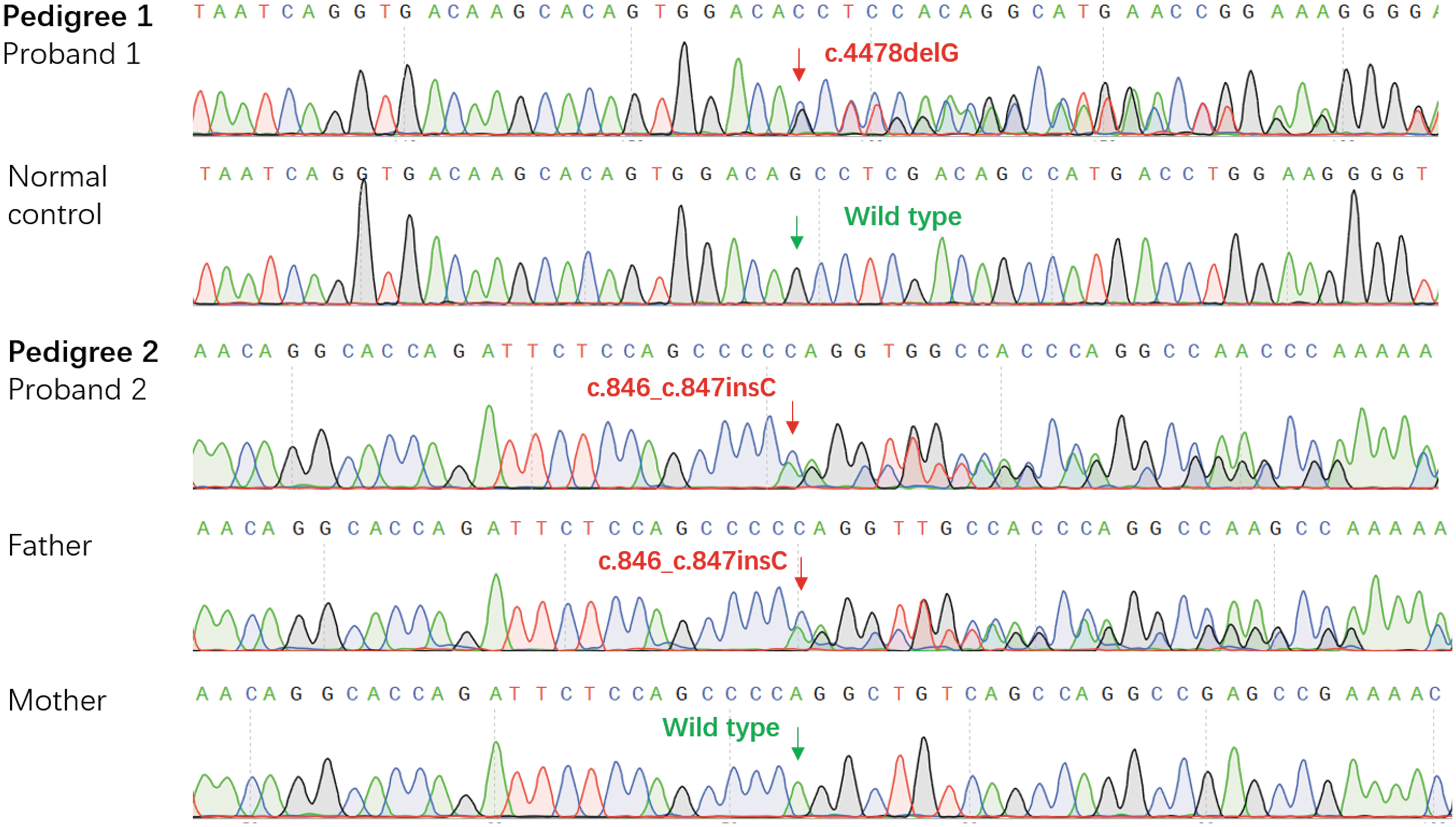
Part sequencing results of the mutated and wild-type fragments from the subjects. The red arrows indicated the mutated sites; the green arrows indicated the wild-type sites.
Variants Associated with Polydactyly Screened by Sanger Sequencing
All the variants in the table were referred to genome hg19.
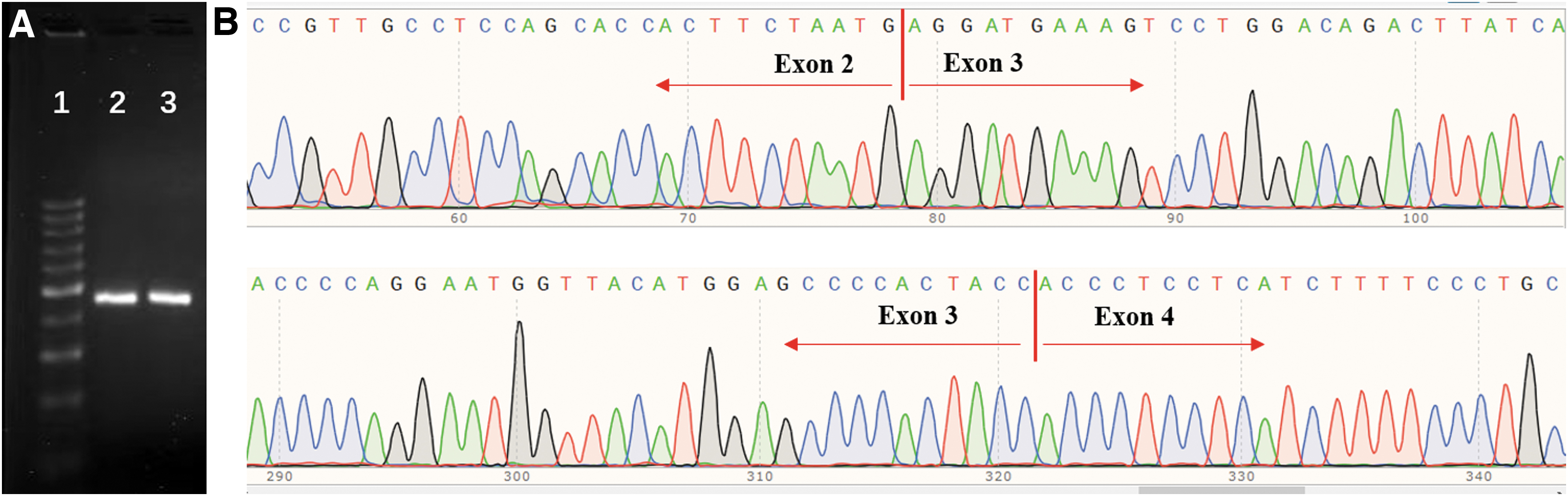
We found that the intronic mutation c.125-47C>A in proband 3 had an uncertain impact on the function of the GLI3 protein. However, we did not detect any mutations in the coding regions of the HOXD13 gene in the three probands. In addition, we identified that the triplet repeats in exon 1 of the HOXD13 gene were normal with 15 GCN (Fig. 3).

Electrophoresis analysis and sequencing of exon 1 in HOXD13 gene from three probands.
Bioinformatics analysis and prediction of mutation severity
According to the ACMG guidelines, we estimated the pathogenic grade of c.4478delG (p.S1493Tfs*18) in the GLI3 gene of proband 1 to be PVS1-Strong (very strong pathogenicity 1-Strong) +PM2 (moderate pathogenicity 2), whereas the pathogenic grade of c.846_c.847insC (p.R283Qfs*21) in the GLI3 gene of proband 2 was estimated to be PVS1+PM2.
Both c.4478delG/(p.S1493Tfs*18 and c.846_c.847insC/p.R283Qfs*21 in the GLI3 gene were not reported in 1000 Genome, GnomAD, or ExAC; however, the frequency of c.125-47C>A was 0.000008 (2/248702) in GnomAD and 0.000025 (3/121080) in ExAC. The ACMG guidelines suggested that the pathogenic grade of this variant was uncertain, and bioinformatics prediction (HSF, spliceAI, and CBS) indicated that it was a deep intron variant that probably had no impact on mRNA splicing.
Splicing effect of c.125-47C>A in GLI3 mRNA and sequencing results of the GLI1 and IQCE genes for proband 3
RT-PCR and sequencing results found that c.125-47C>A of proband 3 had no splicing effect in GLI3 gene (Fig. 5A, B), which was consistent with the prediction of bioinformatics. Moreover, there were no evident mutations found in the GLI1 or IQCE gene by PCR and sequencing.
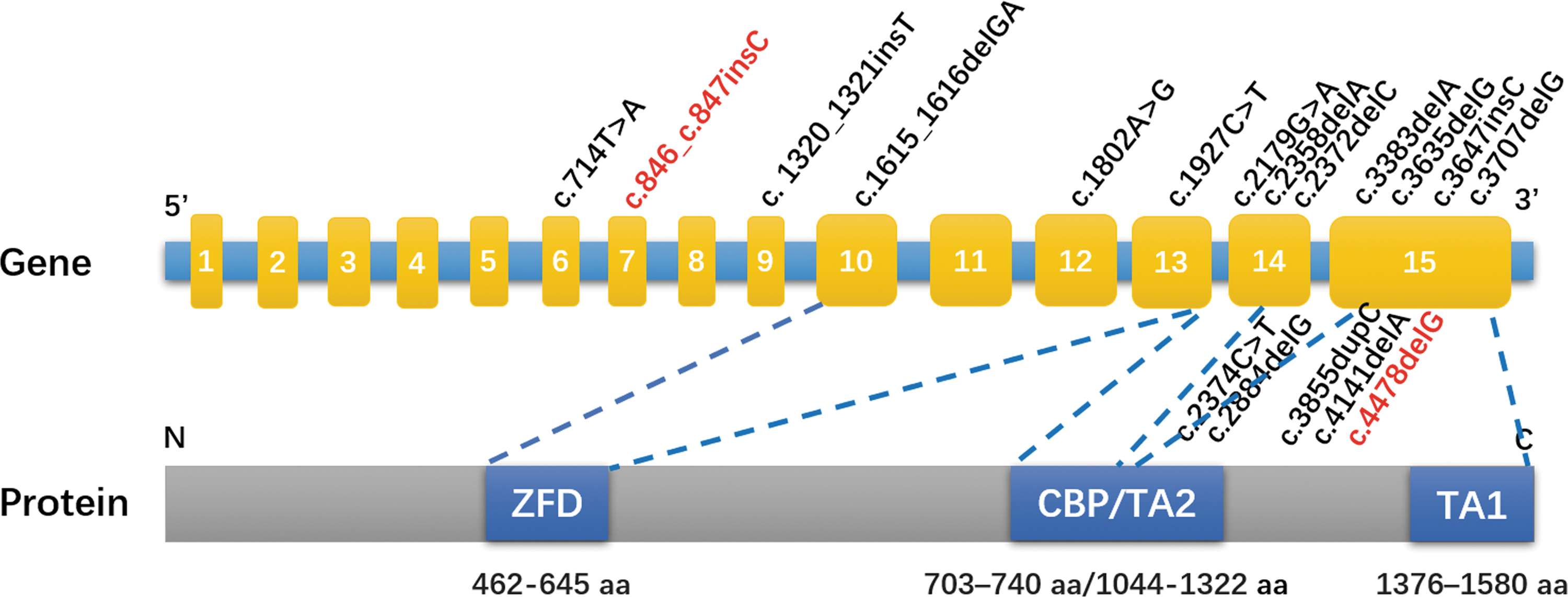
Structural diagram of GLI3 gene and protein, and GLI3 mutations that cause isolated polydactyly. The mutations in red words were novel mutations found in this study, and the mutations in black words were reported in the Human Gene Mutation Database.
RT-PCR and sequencing results of the proband 3.
Discussion
In the current study, we identified that pedigree 1 and pedigree 2 exhibited autosomal dominant heritance of non-syndromic polydactyly, whereas pedigree 3 involved a sporadic case with syndactyly. Interestingly, although the mother in pedigree 1 had PAP, the phenotype of the proband indicated minor variants of SPD (Table 1) (Malik and Grzeschik, 2008).
The proband in pedigree 2 had PPD; however, the phenotype of the father (preaxial-PAP syndactyly complex) was similar to that of a previously reported case limited to the hands, rather than both hands and feet as in our study (Volodarsky et al., 2014). Two novel heterozygous frameshift mutations (c.4478delG/p.S1493Tfs*18 and c.846_c.847insC/p.R283Qfs*21) were identified in the GLI3 gene in pedigree 1 and pedigree 2 (Fig. 2), both of which produced premature stop codons downstream and truncated the GLI3 protein.
The c.846_c.847insC/p.R283Qfs*21 mutation of the GLI3 gene was also verified in the affected father in pedigree 2; however, the unaffected mother carried wild-type alleles at this site (Fig. 2). Therefore, the c.4478delG/p.S1493Tfs*18 mutation of the GLI3 gene in proband 1 was probably transmitted from the affected mother, whereas the c.846_c.847insC/p.R283Qfs*21 mutation in proband 2 was from the affected father.
Interestingly, an intronic variant was found in IVS 2 of the GLI3 gene (c.125-47C>A) in proband 3; nevertheless, supporting evidence from ACMG, bioinformatics prediction (HSF, spliceAI, and CBS), and RT-PCR analysis indicated that this variant was merely a single nucleotide polymorphism with probably no impact (Fig. 5). Moreover, there were no significant mutations identified by PCR-sequencing of the GLI1 or IQCE gene from proband 3.
The GLI3 gene encodes a transcription factor that plays a major role in the canonical Hedgehog signaling pathway (Hui and Angers, 2011), and it functions in tissue patterning and the regulation of limb development. The human GLI3 gene consists of 15 exons and encodes a protein consisting of 1580 amino acids.
The GLI3 protein has several functional domains: transcriptional repressor site at the N-terminus, zinc finger domain, protease cleavage site, CREB binding protein domain (CBP), and two transcriptional activation sites at the C-terminus (TA2 and TA1) (Fig. 4). Mutations at different sites of GLI3 have been closely associated with the varying phenotypes of patients with syndromic polydactyly or non-syndromic polydactyly.
To date, 252 mutations in the GLI3 gene have been reported in the Human Gene Mutation Database (HGMD), with 133 of them being associated with GCPS, and 52 with PHS, 16 mutations with non-syndromic polydactyly was selected and found to be scattered throughout the GLI3 gene, with most (14/16) being located in the 3′ segment with important domains (Fig. 4).
For instance, the c.4478delG/p.S1493Tfs*18is located in the transcriptional activation 1 (TAT1) domain, whereas c.846_c.847insC/p.R283Qfs*21 is found in the N-terminal transcriptional repressor domain, further supporting the observation that GLI3 mutations related to non-syndromic polydactyly are outside of the CBP/TA2 domain (Umair et al., 2019; Wang et al., 2014). Both mutations disrupt the respective domains, and result in the truncation of the protein, affecting its biological function.
SPD is a congenital limb deformity featuring the incomplete separation and duplication in digits, with polydactyly being a common symptom. Of note, most patients with SPD harbor mutations in the HOXD13 gene. Due to the distinctly heterogenous phenotypes and incomplete penetrance of SPD, many patients only have polydactyly or atypical phenotypes in the extremities, so we also amplified and sequenced the HOXD13 gene to ensure that the phenotypes observed in the probands in our study were not partly attributed to the mutations in the HOXD13 gene.
However, due to the coverage of limited hotspot genes of polydactyly, such as GLI3, HOXD13, GLI1, and IQCE genes, our study still could not exclude the involvement of other polydactyly genes such as SHH/ZRS, MIPOL1, ZNF141, and PITX1 in the pathogenicity of malformations in these families.
Conclusions
We reported two novel frameshift and truncated mutations in the GLI3 gene associated with non-syndromic polydactyly in two Chinese families. This study further broadened the mutational spectrum of the GLI3 gene associated with non-syndromic polydactyly, prompting further studies of genotype-phenotype correlations in this disease.
Footnotes
Authors' Contributions
X.G. designed the study and T.S. supervised it. PCR, sequencing analysis, and funding acquisition were performed by X.G. M.L. participated in partial data analysis. B.L. and Y.P. were responsible for disease diagnosis and sample collection. X.G. wrote the article and critically revised it. All authors gave final approval to this paper.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the Science and Technology Plan Project of Fuzhou city (2022-S-017) and Fujian Provincial Clinical Medical Research Center for First Aid and Rehabilitation in Orthopaedic Trauma (2020Y2014).