
Abstract
Background:
Otitis media (OM) is defined as middle ear (ME) inflammation that is usually due to infection. Globally, OM is a leading cause of hearing loss and is the most frequently diagnosed disease in young children. For OM, pediatric patients with Down syndrome (DS) demonstrate higher incidence rates, greater severity, and poorer outcomes. However, to date, no studies have investigated the bacterial profiles of children with DS and OM.
Method:
We aimed to determine if there are differences in composition of bacterial profiles or the relative abundance of individual taxa within the ME and nasopharyngeal (NP) microbiotas of pediatric OM patients with DS (n = 11) compared with those without DS (n = 84). We sequenced the 16S rRNA genes and analyzed the sequence data for diversity indices and relative abundance of individual taxa.
Results:
Individuals with DS demonstrated increased biodiversity in their ME and NP microbiotas. In children with OM, DS was associated with increased biodiversity and higher relative abundance of specific taxa in the ME.
Conclusion:
Our findings suggest that dysbioses in the NP of DS children contributes to their increased susceptibility to OM compared with controls. These findings suggest that DS influences regulation of the mucosal microbiota and contributes to OM pathology.
Introduction
Otitis media (OM), that is, infection and inflammation of the middle ear (ME), is a leading cause of hearing loss globally. In the United States it is the most frequently diagnosed disease in young children, with around half of children experiencing ≥3 episodes of acute (A)OM before the age of 3 years (Collaborators, 2021; Monasta et al., 2012; Teele et al., 1989). OM is often bacterial or viral in origin, wherein pathogens in the nasopharynx (NP) migrate through the Eustachian tube to the ME (Danishyar and Ashurst, 2022; Marom et al., 2012). For instance, multiple viruses can infect the ME and have been identified in samples from MEs with recurrent/acute OM (RAOM), which can then predispose to superimposed bacterial infection that may become recurrent or chronic (Malagutti et al., 2020).
This creates an inflammatory cycle in the ME with an accumulation of mucus and fluid, which can lead to permanent damage and hearing loss (Rosenfeld et al., 2013a). It is important to note that before infection, the ME is essentially sterile (Jervis-Bardy et al., 2019) as it is generally separated from the external environment by the tympanic membrane. In contrast, the NP has an established microbiota that can vary based on microbial exposure and host genetics, but these microbes in the NP do not become resident in the ME if the Eustachian tube is functioning well (Jervis-Bardy et al., 2019). Some NP commensals are potential opportunistic otopathogens of the ME (Yatsyshina et al., 2016). It is well known that increased abundance of potential otopathogens in the NP is associated with higher risk for OM (Browne et al., 2021; Jervis-Bardy et al., 2017; Xu et al., 2021).
In children with Down syndrome (DS), OM occurs at higher rates, with an estimated 68% of children diagnosed with OM with effusion by age 5 years (Barr et al., 2011; Kong et al., 2017; Kreicher et al., 2018; Maris et al., 2014; Omar et al., 2021; Shott et al., 2001). These occurrences are most commonly observed in two age brackets: 1-year-old and 6-7 years old (Maris et al., 2014). Furthermore, OM in patients with DS is typically more severe with worse outcomes. In previous studies, 90% of OM patients with DS required ventilation tubes with an average of 2.39 surgeries per child (Bernardi et al., 2017; Omar et al., 2021). Chronic OM with effusion (COME) is also the leading cause of hearing loss in DS. Repeat tympanostomy tube insertions occur for 61% of children with DS particularly if COME is the indication for surgery (Omar et al., 2021).
The largest potential contributor to these high incidence rates is the altered immune landscape of DS in which immune homeostasis is disrupted and is responsible for many of the clinical phenotypes observed in DS; however, this has not been thoroughly studied for OM (Ram and Chinen, 2011; Waugh et al., 2019). It is well established that individuals with DS have increased susceptibility to infections due to immune factors as well as nonimmune variables. There is also a high rate of Eustachian tube dysfunction, as hypotonia is a common feature of DS, allowing pathogens increased access to the ME as well as preventing the proper drainage of fluid from the ME during infection (Austeng et al., 2013; Chin et al., 2014).
To date, no studies have examined the bacterial profiles of OM in patients with DS. Our hypothesis is that the increased susceptibility to OM in DS is mediated through genetic effects of trisomy 21 such as decreased commensal binding, increased pathogen binding, or reduced clearance of pathogens that alter the microbiota, which in turn lead to OM. For this study, our aim was to examine differences in the NP and ME microbiotas of children with OM according to DS diagnosis.
Materials and Methods
Ethics approval
Ethical approval was obtained from the Colorado Multiple Institutional Review Board before the start of the study. Informed consent was obtained from the parents of children enrolled in the study.
Subject ascertainment
Clinical data were obtained from 105 pediatric patients including 14 with DS, undergoing surgery for OM, with information on age, sex, self-reported ethnicity, family history, breastfeeding history, history of exposure to smoking, OM diagnoses, and surgical technique (Table 1). A total of 330 microbial samples were obtained from the NP (n = 145) and ME (n = 185) of 97 individuals, including 84 ME swabs, 92 ME aspirates, and 7 ME mucosal tissue samples. Four ME cholesteatoma or granuloma tissue samples and 145 NP swabs were also collected. Microbial DNA was isolated from 239 (72%) samples using the Epicentre Masterpure Complete DNA Purification Kit (Lucigen, Middleton, WI); the rest of the samples from which no microbial DNA was isolated were excluded from further study.
Characteristics of Patients With or Without Down Syndrome
RAOM = >3 OM episodes in 6 months or >4 OM episodes in 12 months.
COME = ME effusion persisting for >2 months (Rosenfeld et al., 2013b).
COME, chronic otitis media with effusion; DS, Down syndrome; ME, middle ear; OM, otitis media; RAOM, recurrent/acute otitis media.
Sample collection and 16S rRNA gene sequencing
A total of 185 ME and 145 NP samples were obtained from 97 pediatric patients with OM and submitted for 16S rRNA sequencing. Bacterial profiles were determined by broad-range polymerase chain reaction amplification and sequence analysis of the 16S rRNA gene V1V2 regions, as previously described (Frank et al., 2021; Santos-Cortez et al., 2018). Illumina paired-end sequencing was performed on MiSeq using the 600 cycle version 3 kit. Assembled and quality-filtered sequences were aligned and classified with SINA (1.3.0-r23838) using the 418,497 bacterial sequences in Silva 115NR99 (Pruesse et al., 2012; Quast et al., 2013). Operational taxonomic units (OTUs) were produced by clustering sequences with identical taxonomic assignments (median: 93,598 sequences/sample; interquartile range: 29,447-159,820).
Goods coverage scores (which estimates completeness of sampling) were ≥99.7% for all samples, indicating adequate depth of sequence coverage for all samples. Of the 330 microbial samples submitted for sequencing, 91 did not pass quality control (DNA concentration ≥10 ng/μL; 2500 reads after sequencing). Because it was not possible to determine whether the lack of microbial DNA is due to a relatively sterile ME or from a sample collection issue, these 91 samples were excluded.
Statistical analyses
Bacterial alpha-diversity indices (measuring richness, diversity, and evenness within a sample) were tested for association with DS independently through Wilcoxon test (Robertson et al., 2013). Associations of individual OTUs with DS were assessed using the DESeq2 analysis package in R with sequencing batch as a covariate (Love et al., 2014). To minimize multiple comparisons, only taxa with a prevalence >10% and relative abundance >1% were included in the analysis. Beta-diversity (comparison of biodiversity across samples) was determined through permutational multivariate analysis of variance (PERMANOVA) using the Aitchison dissimilarity index (compares overlap of OTUs between groups) and adjusted for batch effects. R software was used for data analyses and figure generation.
Results
Cohort summary
Microbial samples used in these analyses were collected from 95 pediatric patients with OM with ages ranging from 8.7 months to 14.9 years (median 2.0 years; Table 1). Notably children with DS were significantly older (p = 0.003). In the entire cohort and in each subset analyses, males were predominant, which is a known phenomenon for OM (Paradise et al., 1997); however, there were no significant sex differences between those with or without DS. Of note, the rate of chronic OM was higher in patients with DS (p = 0.005) compared with non-DS patients for whom RAOM was a more common diagnosis. In addition, variables that were significantly different in DS versus non-DS were type of OM surgery and antibiotic history (Table 1), both of which are partly determined by the OM diagnosis. As expected, due to the disease mechanism for DS during meiosis (de novo trisomy), family history was different (p = 0.03), with 52.4% of non-DS children having a family history of OM (Elling et al., 2021).
ME microbiota
A total of 330 microbial samples were initially collected from the NP and ME of 97 children. For microbiota analyses, samples were filtered for (1) those with >2500 16S rRNA sequencing reads and (2) one ME and one NP sample per individual where bilateral samples were collected (if bilateral, right-sided sample was used). No differences were identified between right and left NP or ME samples from the same individuals in PERMANOVA and principal component analyses (data not shown). After filtering, 16S rRNA sequence data from 47 ME and 91 NP samples from 95 children were analyzed according to DS diagnosis (Table 1).
ME samples of OM patients with DS demonstrated greater diversity and evenness (Fig. 1) with significant differences in alpha-diversity indices Shannon's diversity index (quantitative measure of richness; p = 0.007) and Shannon's equitability (quantitative measure of evenness; p = 0.002). Overall microbiota composition was significantly different between OM patients with and without DS as determined by PERMANOVA analyses including adjustment for batch effects (Fig. 2; Supplementary Fig. S1).
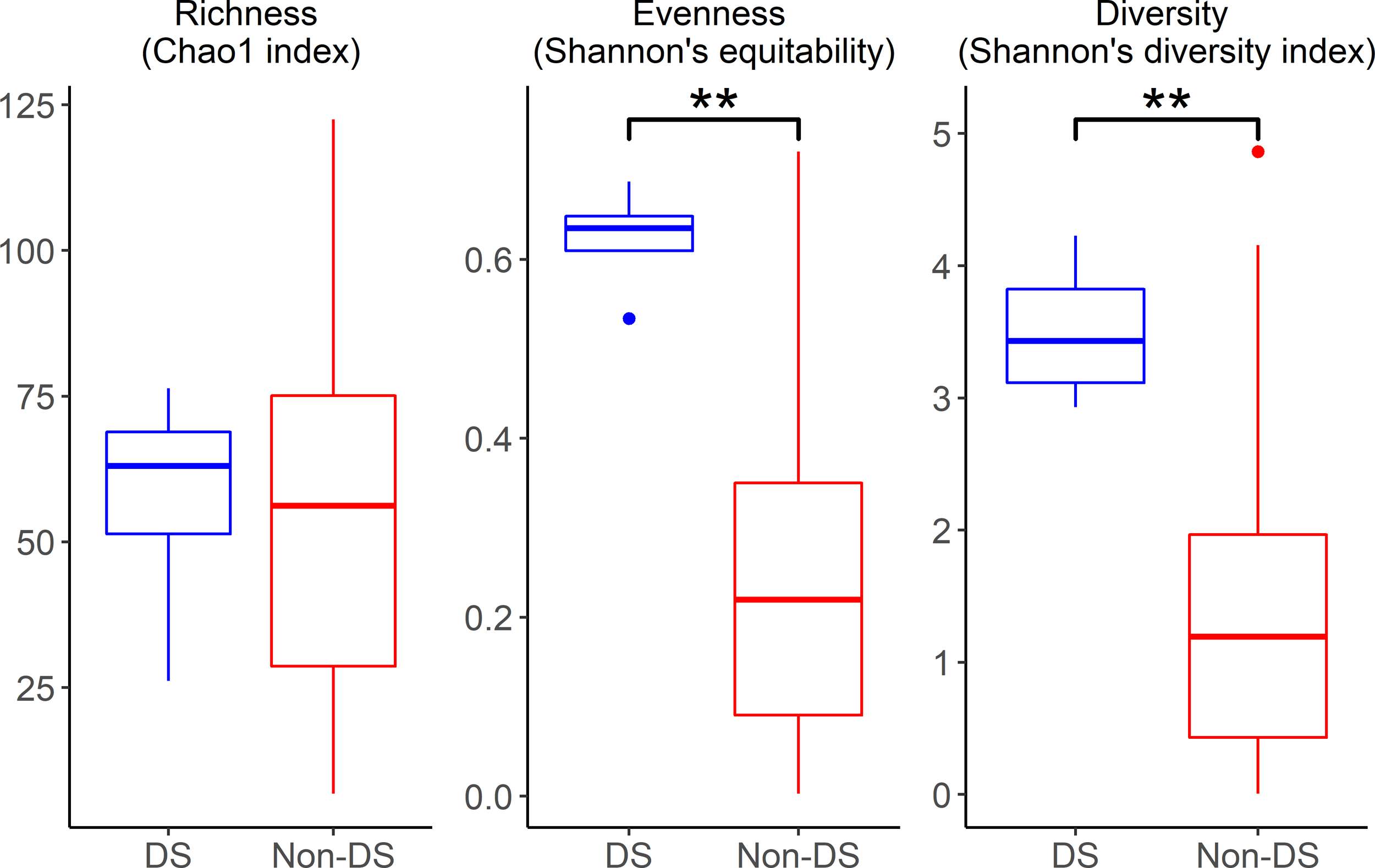
Alpha-diversity indices of ME samples demonstrate significantly increased biodiversity in OM patients with DS, namely in evenness (Shannon's equitability; p = 0.002) and diversity (Shannon's diversity index; p = 0.007). **p-Value <0.01. DS, Down syndrome; ME, middle ear; OM, otitis media.
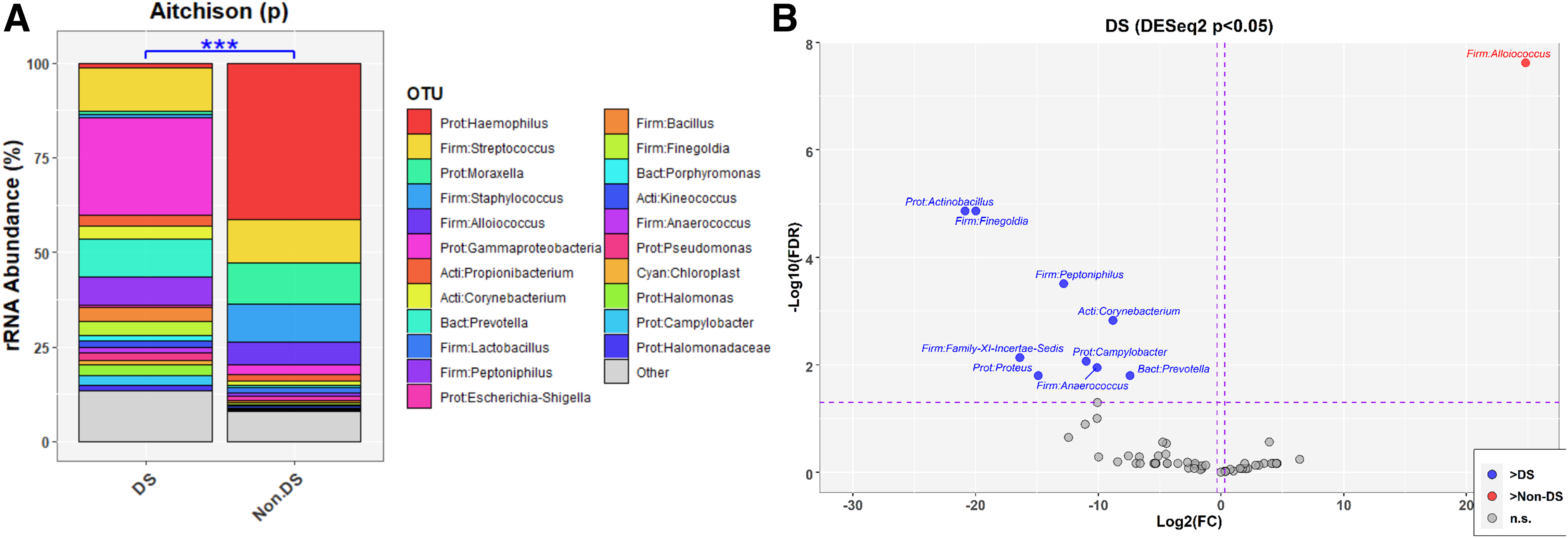
Several taxa showed a significant increase in relative abundance in the ME of OM patients with DS: Actinobacillus (false discovery rate adjusted or FDR-p = 1.4×10−5), Corynebacterium (FDR-p = 0.001), Prevotella (FDR-p = 0.02), Anaerococcus (FDR-p = 0.01), Family-XI-Incertae-Sedis (FDR-p = 0.007), Peptoniphilus (FDR-p = 0.0003), Proteus (FDR-p = 0.02), Finegoldia (FDR-p = 1.4×10−5) and Campylobacter (FDR-p = 0.009). Meanwhile, Alloiococcus (FDR-p = 2.4×10−8) was enriched in OM patients without DS (n = 44) as compared to OM patients with DS Fig. 2B).
These ME microbiota analyses were repeated independent of DS using either OM type or age as the classifier. In the ME, there were no significant differences in alpha-diversity indices by either age (Supplementary Fig. S2) or OM type (Supplementary Fig. S3). Examination of changes in relative abundance of individual taxa by age or OM type largely reflected what is known about these taxa and their respective roles in RAOM or COME (Supplementary Figs. S4 and S5) and did not recapitulate taxa findings in the ME based on DS status (Fig. 2). Taken together, these additional analyses support the findings that the increased biodiversity and greater relative abundance of several taxa in the MEs of individuals with DS were not due to differences in age or OM type.
Nasopharyngeal microbiota
Similar to the ME, NP samples of OM patients with DS demonstrated greater diversity and evenness (Fig. 3), with significant differences in alpha-diversity indices Chao1 (bacterial richness; p = 0.003), Shannon's diversity index (p = 0.002), and Shannon's equitability (p = 0.01). Furthermore, there was a significant difference (p < 0.01) in beta-diversity after correcting for age and batch (Fig. 4A; Supplementary Fig. S6). Age, sex, and OM type were significant covariates in this model and thus not included. Similar to the ME, there were significant differences in beta-diversity after correcting for batch (Fig. 4; Supplementary Fig. S6).

Alpha-diversity indices of NP samples demonstrate significantly increased biodiversity in OM patients with DS in evenness (Shannon's equitability; p = 0.01), diversity (Shannon's diversity index; p = 0.002), and richness (Chao1; p = 0.003). **p-Value <0.01. NP.
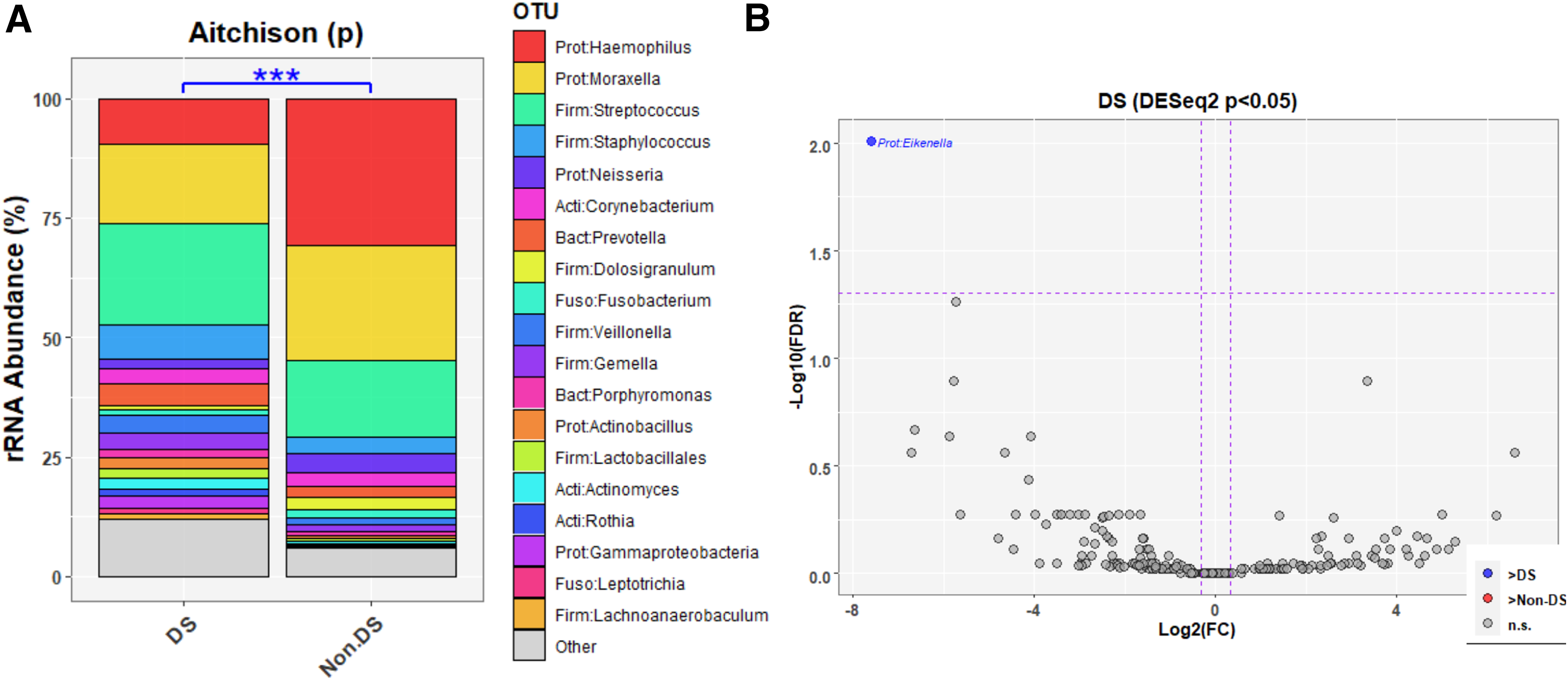
In the NP, Eikenella had significantly increased relative abundance in individuals with DS (FDR-adjusted p = 0.01) (Fig. 4).
In the NP, Chao1, Shannon's equitability and Shannon's diversity indices were all significantly greater in patients >5 years old as compared with those <5 years (Supplementary Fig. S7), although there were no significant differences in alpha-diversity indices by OM type (Supplementary Fig. S8). As in the ME, bacterial profiles by age (Supplementary Fig. S9) or OM type (Supplementary Fig. S10) were very different compared to profiles by DS status. Like in the ME, these NP findings indicate that the microbiota differences according to DS status were not due to age or OM type. It should be noted that beta-diversity was not significant in DS versus non-DS after correction for age (data not shown). As in the ME, bacterial profiles by age (Supplementary Fig. S9) or OM type (Supplementary Fig. S10) were very different compared with profiles by DS status, such that Eikenella was the only taxon that was significantly increased in NPs of individuals with DS. In contrast, Eikenella was not identified in the NP microbiota profiling based on age or OM type, further suggesting that the NP findings in individuals with DS were not due to differences in age or OM type.
The ME and NP analyses were repeated with age- and sex-matched individuals (Supplementary Table S1). In the ME, biodiversity was not significantly different, likely due to decreased power because of the smaller sample size after matching (Supplementary Figs. S11 and S12A). The analyses of relative taxa abundance recapitulated much of the aforementioned findings, including Finegoldia, Peptoniphilus, and Family-XI-Incertae-Sedis identified as significantly enriched in OM patients with DS reinforcing that these microbiota differences were potentially due to DS rather than age differences (Supplementary Fig. S12B).
In the NP, biodiversity was increased in DS samples as measured by both alpha-diversity indices and beta-diversity (Supplementary Figs. S13 and S14A). In the NP, Halomonas and Halomonadaceae were both enriched in DS individuals, as was also observed in the ME analysis utilizing age- and sex-matched individuals (Supplementary Fig. S14B), whereas Peptostreptococcaceae and Cardiobacteriaceae were enriched in controls.
Discussion
In pediatric OM patients with DS, there were increases in microbiota bio-diversity and an enrichment of multiple taxa in both the NP and ME as compared with pediatric OM patients without DS. Of the taxa identified as being significantly enriched in the ME microbiota in individuals with DS and OM, Prevotella, Anaerococcus, Peptoniphilus, Corynebacterium, Proteus and Finegoldia as well as Eikenella in the NP, have all been previously identified in connection with OM (Supplementary Fig. S11; Ari et al., 2019; Boers et al., 2018; Man et al., 2019; Walker et al., 2019; Oishi et al., 2021; Tesfa et al., 2020). Furthermore, Eikenella, Prevotella, Corynebacterium, Anaerococcus, Peptoniphilus and Campylobacter have all been identified as NP colonizers or commensals (Oishi et al., 2021; Klein et al., 1990; Rosas-Salazar et al., 2021). These findings suggest that although the taxa that make up the OM microbiotas of patients with DS are not unique to DS, these taxa altogether form a dysbiotic composition that differs from OM patients without DS. Notably, there was a significant increase in relative abundance of Halomonas in both NP and ME associated with DS when performing age- and sex-matched analyses. Halomonas is a halophilic gram-negative proteobacteria that includes several species determined to be human pathogens (Stevens et al., 2009). Furthermore, Halomonas was a dominant taxon present in ME effusions from a cohort of pediatric patients with OME, further supporting its role as a potential otopathogen (Xu et al., 2020). In addition, Halomonas was identified to abundantly co-occur with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in the NP and oral cavity in patients with subclinical inflammation (Acharya et al., 2017; Mehta et al., 2021). More studies concerning the role of Halomonas in OM are needed to determine its potential role as an otopathogen.
Our DS cohort largely reflects what is observed in the literature with regard to DS children as well as DS children with OM. Our DS cohort had a higher median age than our non-DS cohort and, as previously mentioned, this is due to the second, older pediatric age bracket at an average of 6-7 years old in which OM is observed in children with DS, whereas in non-DS children, most OM cases are in children under the age of 3 years (Maris et al., 2014). Furthermore, in this study COME was the predominant OM type in children with DS (Table 1), reflective of what is observed in the literature (Chin et al., 2014). This is not surprising given that chronic infections are more commonly observed in DS as a result of altered immune homeostasis (Ram and Chinen, 2011; Waugh et al., 2019).
Other potentially relevant observations include the lower (although nonsignificant) observed rate of breastfeeding in DS individuals with OM in our cohort as compared with non-DS. Although difficulty of breastfeeding is not uncommon in DS infants who have poor suck due to hypotonia and dysphagia, lack of breastfeeding could lead to deviations in the development of their immune system and oral microbiotas from those who are breastfed (Laouar, 2020; Ogra and Welliver, 2008; Pisacane et al., 2003; Stanley et al., 2019). In addition, there was a lower incidence of family history of OM documented in DS individuals from our cohort despite the moderate heritability of OM in general (Casselbrant et al., 1999). This supports our hypothesis that OM susceptibility in DS is directly related to the genetic architecture of DS due to de novo trisomy in the child and its downstream effects.
Individuals with DS are at an increased risk of periodontal disease, in which the oral microbiota plays a large role as it is defined by sustained infection and inflammation of the gingiva (Scalioni et al., 2018). In addition, several studies have shown that there is a difference in the normal microbiotas of those with DS as compared with those without, including an increase in the load of periodontitis-related pathogens (Vocale et al., 2021; Willis et al., 2020).
Many of the nominally significant taxa identified in our study had overlap with periodontitis and/or the oral microbiota of individuals with DS in the literature (Mitsuhata et al., 2022; Supplementary Fig. S15). None of the taxa were identified in association with DS exclusively, likely due to the scarcity of microbiome studies for DS as well as the high prevalence of periodontitis in patients with DS, estimated to be 58-96% (Morgan, 2007). This might suggest that the oral microbiota of individuals with DS is a potential reservoir for otopathogens that contribute to the increased occurrence of OM in children with DS.
Study limitations include the small sample size, which is partly due to the limited DS population undergoing OM surgery particularly during the SARS-CoV-2 pandemic and also restricted access to ME samples; the cross-sectional study design; lack of information on viral and immune profiles in the NP and ME; and the inability to identify bacterial species, which is an inherent limitation of the methods used for bacterial profiling. This research is important as a bacterial study given that samples were collected at the height of the SARS-CoV-2 pandemic when utilization of health services for OM was low. Nonetheless, we were able to identify significant differences even with correction for multiple testing of individual taxa, which may indicate the strength of associations identified.
Moreover, these findings may guide the use of antibiotics for OM in children with DS, for example, expanding antibiotic coverage to target gram-negative bacteria (Yeo et al., 2016). Additional research that includes longitudinal follow-up of microbiota in a larger cohort of children, species identification and antibiotic sensitivity profiling for abundant taxa, and co-occurring immune and microbiota profiles in the NP and ME will help elucidate the disease mechanisms behind the associations between DS and mucosal microbiota profiles that were identified in this study and aid in the selection of antibiotics for OM treatment.
Conclusion
To summarize, we identified differences in the NP and ME microbiotas of children with OM according to DS status, including increased biodiversity and greater relative abundance of several taxa with DS.
Footnotes
Acknowledgments
The authors would like to thank the patients who provided clinical data and samples; S. Streubel, P. Kelley, S. Cass, and S. Gubbels for their help with sample collections; and T. Bootpetch for microbial DNA isolation.
Authors' Contributions
Conceptualization, supervision, project administration and validation by D.N.F., B.W.H., and R.S.C. Funding acquisition by C.L.E., B.W.H., and R.S.C. Methodology, resources, and investigation by C.L.E., S.H.G., S.D.H., K.T., J.M.K., D.C., C.E.R., J.D.P., P.J.Y., T.M.W., K.H.C., M.A.S., N.R.F., D.N.F., B.W.H., and R.S.C. Software by C.L.E., C.E.R., and D.N.F. Data curation by D.N.F. and R.S.C. Formal analysis, visualization, and writing—original draft preparation by C.L.E. Writing—review and editing by C.L.E., D.N.F., B.W.H., and R.S.C. All authors read, critically appraised, and approved the article.
Data Availability Statement
Demultiplexed 16S rRNA paired-end sequence data and associated metadata were deposited in the NCBI Sequence Read Archive under Bioproject ID PRJNA748418.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
C.L.E. was supported by the National Institutes of Health (NIH)—National Institute on Deafness and Other Communication Disorders (NIDCD), grant number T32 DC012280 (to Sue C. Kinnamon and Herman A. Jenkins). This research was funded by the NIH—NIDCD through grant R01 DC015004 (to R.S.C.), and by the Ponzio Research Accelerator Award from the Center for Children's Surgery, Children's Hospital Colorado (to B.W.H.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.