
Abstract
Background:
Wolfram syndrome (WFS) is an autosomal recessive disorder that often leads to diabetes, optic atrophy, and sensorineural hearing loss. The aim of this study was to determine the clinical characteristics and the genetic cause of the first two Moroccan families presenting with WFS.
Methods:
The clinical features of five members of two WFS families were evaluated. Whole-exome sequencing was conducted to explore the underlying genetic cause in the affected patients.
Results:
Two homozygous variants in the WFS1 gene were identified, each in one of the two families studied: a missense c.1329C>G variant (p.Ser443Arg) and a nonsense mutation c.1113G>A (p.Trp371Ter). These variants affected conserved amino acid residues, segregated well in the two families, and are absent from genetic databases and in controls of Moroccan origin. Bioinformatics analysis classified the two variants as pathogenic by in silico tools and molecular modeling.
Conclusion:
Our study identified for the first time two variants in Moroccan patients with WFS that extends the mutational spectrum associated with the disease.
Introduction
Wolfram syndrome (WFS) (MIM 222300), also known as DIDMOAD (Diabetes Insipidus, Diabetes Mellitus, Optic Atrophy, and Deafness), is a rare and progressive genetic disorder with a significant impact on multiple organ systems. This autosomal recessive disorder is characterized by the development of early-onset diabetes mellitus, diabetes insipidus, optic atrophy, and sensorineural hearing loss. Clinical features of WFS extend beyond the hallmark symptoms, often encompassing additional complications such as neurological abnormalities, psychiatric disorders, urinary tract abnormalities, and other endocrine dysfunctions (Serbis et al., 2023). The progressive nature of the syndrome poses significant challenges for patients and their families, warranting a multidisciplinary approach to management and care (Pallotta et al., 2019), especially since the prognosis of the disease is poor, leading to brainstem atrophy and respiratory failure ultimately resulting in premature death, usually occurring in the fourth decade of life (Png et al., 2023).
Among the underlying causes of WFS are mutations in the WFS1 gene located on chromosome 4p16.1, which spans approximately 30 kilobases and consists of 8 exons encoding the wolframin protein (Polymeropoulos et al., 1994; Strom et al., 1998). Wolframin is primarily expressed in the endoplasmic reticulum (ER) of various tissues and is involved in the regulation of ER calcium homeostasis and the unfolded protein response. Dysfunction of wolframin leads to ER stress, impaired cellular function, and eventual apoptosis in multiple target tissues, contributing to the clinical manifestations observed in WFS (Serbis et al., 2023).
WFS is evaluated to affect between 1 in 160,000 and 770,000 people worldwide (Urano, 2016). However, accurate estimations are challenging due to the frequent misdiagnosis of WFS as type 1 diabetes during its early stages (Wang et al., 2019), and genetic analysis is often necessary to confirm the diagnosis.
To date, over 400 different mutations in the WFS1 gene have been reported in patients with WFS. These mutations exhibit a wide range of distribution throughout the gene, with a significant concentration in the largest exon 8, particularly in the region encoding the transmembrane and C-terminal domain of the protein (Pallotta et al., 2019; Zhang et al., 2022). These mutations can include missense, nonsense, frameshift, and splice site mutations, as well as large-scale deletions and insertions. The majority of WFS1 mutations result in premature termination of protein synthesis or impair the normal function of the wolframin protein (Astuti et al., 2017; Cryns et al., 2003; Rigoli et al., 2022). The diverse nature of these mutations contributes to the phenotypic heterogeneity observed in WFS, with varying degrees of severity and clinical manifestations (Serbis et al., 2023).
Herein, we report five cases belonging to two consanguineous families previously undiagnosed, presenting with progressive vision loss, type 1 diabetes, and neurological manifestations. Whole exome sequencing (WES) identified novel homozygous variants in the exon 8 of WFS1, classified pathogenic by in silico tools and molecular modeling, confirming the WFS.
Materials and Methods
Patients recruitment
Five patients belonging to two families were referred to the department of Neurology, Specialties Hospital (Rabat, Morocco) for etiological assessment of diabetes, visual loss, epilepsy, and movement disorders. Parents of the two families were healthy and second-degree relatives, assuming an inherited disease with autosomal recessive mode of inheritance. Signed informed consent was obtained from all subjects or their legal guardians for these studies in accordance with the Helsinki principles, and the study protocol was approved by the local ethics committee of Cheikh Zaid Hospital (approval number: CEFCZ/PR277).
Genetic analysis
Blood samples were collected from all patients and their parents. Genomic DNA was extracted using the Wizard® Genomic DNA Purification Kit A1120 (Promega). The whole exome sequencing (WES) was performed on the index case of the two families (IV.5 and V.2, respectively), using Ion Proton System and Ion Chef instrument (Thermo Fisher Scientific) as previously described (Bouhouche et al., 2021). The variants identified were confirmed by Sanger sequencing using SeqStudioTM Genetic Analyzer (Thermo Fisher Scientific).
Bioinformatics analysis
The Variant Effect Predictor (VEP) web server was used for analyzing and predicting the impact of genetic variants on protein function. It incorporates a range of established algorithms to calculate scores and provide predictions for different mutation types. Some of the prominent tools integrated into VEP include SIFT, PolyPhen, Proven, LRT, MutationTaster, FATHMM-MKL, and BayesDel. To assess the potential impact of the p.Ser443Arg amino acid change on the WFS1 protein structure, a molecular modeling analysis was conducted. The I-TASSER web server was used to predict the three-dimensional structure for the WFS1 protein. Then, 3D structures of both native and mutated proteins were refined through energy minimization using the Yasara Energy Minimization server. The YASARA View software was used for the analysis of amino acid interactions. Finally, three computational tools, namely DUET, CUPSAT, and DeepDDG, were used to evaluate the impact of the amino acid change on protein stability.
Results
Clinical finding
The pedigrees of the two second-degree consanguineous families of Moroccan origin, comprising respectively three and two affected siblings with suspected WFS, are presented in Figure 1a and d, and the clinical data of all patients are summarized in Table 1.
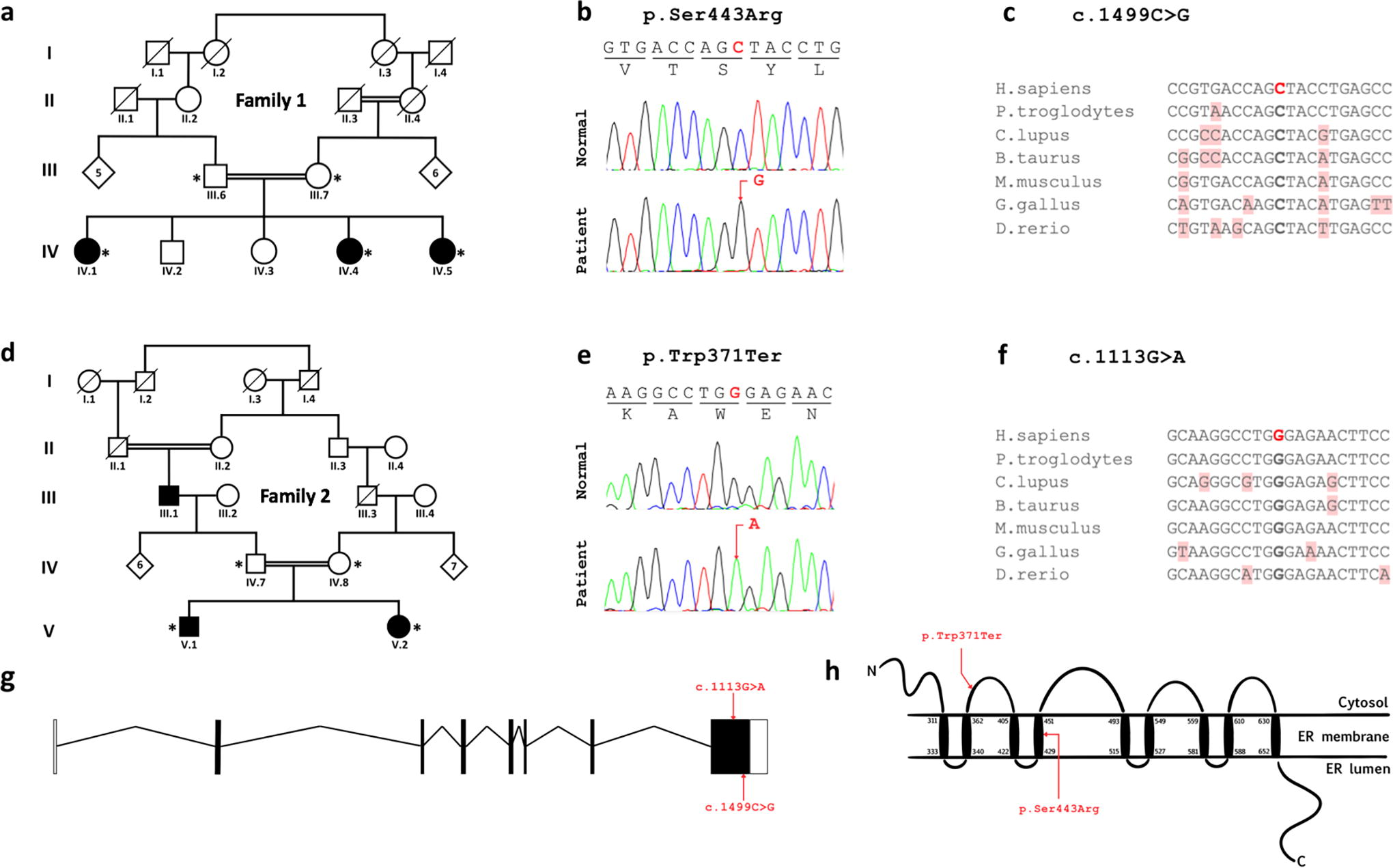
Pedigrees of the two families reported in this study, and the location of disease-associated WFS1 variants on gene and protein levels.
Clinical Features Patients from the Two Studied Families with Wolfram Syndrome
Family 1: The patient IV.1 is a 39-year-old female suffering from type 1 diabetes diagnosed at 18 months, severe hearing loss and progressive loss of visual acuity since the age of 8 ultimately resulting in blindness. Examination confirmed that she also suffers from memory impairments and some degree of dysarthria without ataxia. The patient IV.4 is a 25-year-old suffering from type 1 diabetes since the age of 3, and from a hearing loss followed by progressive loss of visual acuity at age 7 causing her to see only shadows and white. Her neurological examination was unremarkable except for some working memory impairments as she can only retain one instruction at a time. Their sister IV.5 is a 23-year-old female suffering from type 1 diabetes, hearing loss and progressive visual impairment since the age of 7. At the age of 16, she started experiencing generalized tonic-clonic seizures, which were then controlled with valproate, as well as a left upper limb postural tremor that improved upon propranolol administration. However, the patient still reported some stimuli-sensitive myoclonus, groaning, and snoring while sleeping. Upon neurological examination, she was found to sustain a dysarthria with no gait or limb ataxia and had normal motor, reflex, and sensory findings. The ophthalmological examination identified the presence of an optic atrophy. The audiogram revealed a bilateral sensorineural hearing loss, and the EEG was normal. The father III.7, aged 64, is asymptomatic with unremarkable family history, whereas the mother III.6, aged 58, is diabetic and has been suffering from high blood pressure since the age of 47.
Family 2: The patient V.1 is a 14-year-old boy with autistic spectrum disorder. He suffers from hallucinations and optic atrophy. He also presents facial dysmorphy with little ears and ligaments hyperlaxity, especially in the elbow. His MRI was unremarkable except for a bilateral optic atrophy. His sister V.2 is an 11-year-old girl presenting with type 1 diabetes, diagnosed at 6 years old, having received 30 UI of intermediate-acting insulin and 20 UI of rapid-acting insulin, and suffering from language acquisition delay, bilateral optic neuropathy that rapidly evolved into optic atrophy, and visual and auditory hallucinations. She was diagnosed with autistic spectrum disorder at age 7. Her MRI showed a bilateral optic nerve atrophy. The EEG and metabolic exploration showed normal results, especially for lactate, creatine-phosphokinase, and homocysteine.
Genetics
Analysis of WES data by Ion Repoter software of patients IV.5 from family 1 and V.2 from family 2 yielded a total of 38913 and 39575 variants, respectively. The obtained variants were then filtered with strict criteria of functional scores of SIFT and Polyphen, variant classification, variant effect, allele frequency, a global MAF <0.001, and under an autosomal recessive model of inheritance. A variant in exon 8 of WFS1 in the homozygous state was identified in patient IV.5 of family 1, consisting of the missense mutation c.1329C>G, which replaces the amino acid Serine by the Arginine at position 443 (p.Ser443Arg). In patient V.2 of family 2, a homozygous nonsense mutation, also in exon 8 of WFS1, was identified, replacing the nucleotide guanine with an adenine at position 1113 (c.1113G>A). This substitution leads to a premature stop codon (p.Trp371Ter), truncating the last 520 amino acids of the protein. Sanger sequencing of WFS1 exon 8 validated the two variants (Fig. 1b and e) and revealed that all the patients were homozygous and the parents of the two families were heterozygous for the respective variant. Moreover, the p.Ser443Arg variant has not been previously reported in dbSNP, 1000 Genomes, gnomAD, ClinVar and in our in-house WES databases, whereas the p.Trp371Ter was reported once in gnomAD Exomes database (rs1239399323) at heterozygous state. They could be considered therefore as novel variants. Alignment of WFS1 sequences in several species showed high conservation of the two mutated nucleotides and thus the corresponding amino acids 371Try and Ser443 (Fig. 1c and f). These two mutated amino acids are located in the cytosol and the forth transmembrane domains, respectively (Fig. 1h).
Bioinformatics
The nonsense mutation p.Trp371Ter was predicted to be deleterious based on four bioinformatics algorithms, including LRT, MutationTaster, FATHMM-MKL, and BayesDel. Similarly, multiple prediction tools (SIFT, Polyphen, Proven, LRT, MutationTaster, FATHMM-MKL, and BayesDel) classified the missense mutation p.Ser443Arg as pathogenic (Table 2).
Computational Analysis of the Impact of the Identified Mutations in the WFS1 Gene
Based on molecular modeling analysis, the p.Ser443Arg mutation might slightly disrupt the interactions between the WFS1 protein amino acids. It may lead to the loss of a hydrogen bond between the amino acids Ser443 and Phe439. In the native structure, there are two bonds between these two residues, but in the mutated structure, there is only one bond (Fig. 2). The analysis of protein stability showed that the p. Ser443Arg substitution potentially induces destabilization in the 3D structure of the WFS1 protein, as predicted by DUET, CUPSAT, and DeepDDG tools (Table 3). Consequently, according to this bioinformatics assessment, the p.Ser443Arg substitution may impact the structure and function of the WFS1 protein.
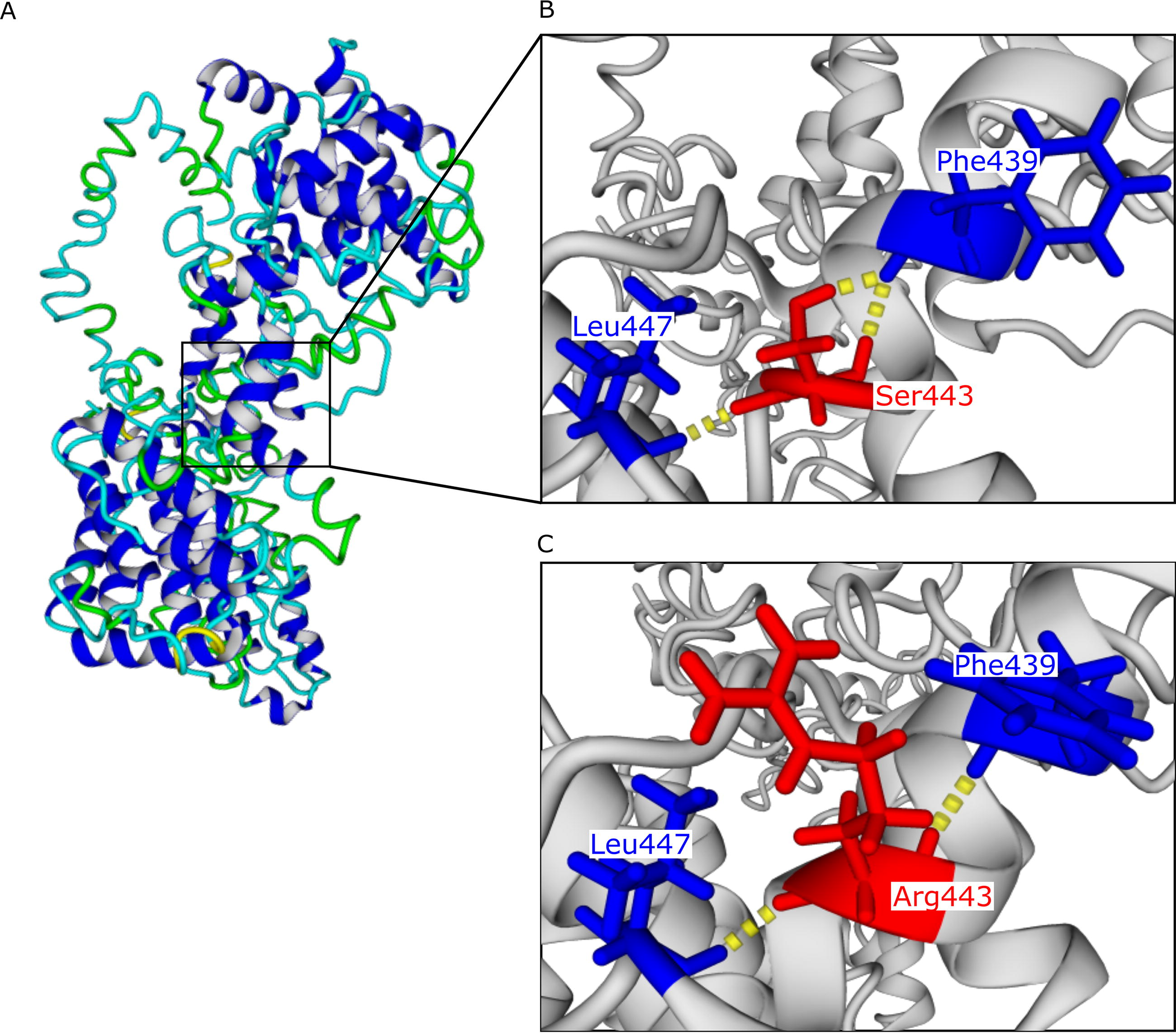
Prediction of the structural impact of the p.Ser443Arg mutation.
Prediction of the Amino Acid Change on the Protein Stability
Discussion
In this study, we present the clinical and genetic findings of two families of Moroccan origin presenting with a multisystem disorder. The patients exhibited a wide range of symptoms, initially leading to their referral across different departments until their neurological abnormalities prompted evaluation in the neurology department. Following a thorough clinical investigation, WFS was suspected and subsequently confirmed through genetic analysis. Notably, these cases represent the first documented instances of WFS in Morocco.
WFS manifested at an early age in all of our patients, although its diagnosis was delayed until decades later for some cases. This highlights the challenges associated with recognizing this rare disorder, which can lead to a delay in implementing appropriate management strategies. In the first family, the eldest sister (IV.1) is entering the fourth decade of her life, a critical period where respiratory complications often arise and contribute to mortality. Currently, she does not exhibit respiratory problems; however, close monitoring is essential due to the high risk associated with this stage of the disease.
Loss of vision, primarily due to optic atrophy, emerged as the most prevalent symptom among the affected individuals in our families. All five patients presented with visual impairment, highlighting the significant impact of optic atrophy in WFS. Diabetes, another hallmark feature of WFS, was observed in all patients except for V.1, who had a late onset at the age of 9. Given the relatively recent onset of symptoms and the short time since their appearance, it is possible that not all symptoms have fully manifested in this patient. Hearing loss was the third most common manifestation, observed in all three sisters of the first family. Other symptoms were less consistently presented, occurring in one or two patients at a time. Psychotic and neurological abnormalities observed in our cases align with the established symptomatology of WFS. Previous studies have reported Autism Spectrum Disorder (ASD) as a clinical feature of WFS (Bischoff et al., 2015), while other patients have exhibited dysmorphic features (El-Shanti et al., 2000). In the large series by Barrett et al. (1995), myoclonus was observed in 62% of cases, mainly during the fourth decade, while it was only present in one patient from the two families studied here, since the cases were still young. Dysarthria is more commonly reported as part of cerebellar ataxia, whereas tremor has not been reported in other series. Cognitive impairment was not consistently reported in the literature. It was even refuted by the study of Bischoff et al. (2015) where they compared patients with confirmed WFS mutation with age- and gender-matched diabetic and nondiabetic controls. The delayed language acquisition in one of our patients was not linked to deafness, which makes it another symptom of this genetic syndrome. Further investigations involving additional cases with the identified variants are essential to explore potential genotype-phenotype correlations and expand our understanding of the clinical heterogeneity observed within WFS.
WES of the affected individuals revealed the presence of two distinct variants in the WFS1 gene, both in the exon eight. The family 1 exhibited a missense mutation, specifically c.1499C>G (p.Ser443Arg), while the family 2 carried a nonsense mutation, c.1113G>A (p.Trp371Ter). The novel missense p.Ser443Arg mutation, identified in the transmembrane region of the protein, has been classified as pathogenic based on predictive tools such as SIFT and Polyphen, and is likely to impact the structure and function of the WFS1 protein as shown by bioinformatics modeling. Whereas the nonsense p.Trp371Ter mutations typically result in premature codon termination, leading to the production of truncated and nonfunctional protein. This mutation was reported once in gnomAD Exomes database (rs1239399323) and described by Bessahraoui et al. (2014) in a family originating from the Oran region in western Algeria. Our second Moroccan family with this same mutation comes from the city of Nador in the north-east of Morocco, which has a border with the Wilaya of Oran. This suggests that the p.Trp371Ter could be a founder effect mutation, originating from this region of North Africa. Additionally, both identified mutations affect conserved nucleotides in WFS1 across the seven species compared. The presence of novel mutations in these Moroccan families suggests the possibility of ethnic-specific variants within the WFS1 gene.
Conclusion
In summary, we identified for the first time in the Moroccan population two variants in WFS1, a nonsense p.Trp371Ter and a novel missense Ser443Arg, both located in exon 8, that are associated with autosomal recessive WFS in five patients belonging to two inbred families. The p.Trp371Ter variant results in the production of a truncated, non-functional protein, whereas the Ser443Arg variant was classified pathological since it could impact the structure and function of the WFS1 protein as shown by molecular modeling. The genetic diagnosis of the patients studied was useful for therapeutic management and for the genetic counseling of the families.
Footnotes
Acknowledgment
The authors thank the patients and their family members for their collaboration in this study.
Authors’ Contributions
Ah.B. designed the study and responsible for the organization and the management of the study. T.H., W.B., Al.B., F.-Z.O. and W.R. evaluated the clinical phenotype. Ah.B., N.B. and F.-Z.E. performed the experiments. H.C. did the bioinformatic analysis. S.S., H.C. and Ah.B. wrote the original draft. Ah.B. and W.R. edited and revised the article. All authors approved the submitted version of the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the “Centre National de Recherche Scientifique et Technique” (CNRST) and the Mohammed V University in Rabat (UM5R), Morocco.