
Abstract
Background:
Hereditary nonsyndromic hearing loss (NSHL) is an extremely heterogeneous disorder, both genetically and clinically. Myosin VI (MYO6) pathogenic variations have been reported to cause both prelingual and postlingual forms of NSHL. Postlingual autosomal dominant cases are often overlooked for genetic etiology in clinical setups. In this study, we used next-generation sequencing (NGS)-based targeted deafness gene panel assay to identify the cause of postlingual hearing loss in an Indian family.
Methods:
The proband and his father from a multigenerational Indian family affected by postlingual hearing loss were examined via targeted capture of 129 deafness genes, after excluding gap junction protein beta 2 (GJB2) pathogenic variants by Sanger sequencing. NGS data analysis and co-segregation of the candidate variants in the family were carried out. The variant effect was predicted by in silico tools and interpreted following American College of Medical Genetics and Genomics-Association for Molecular Pathology guidelines.
Results:
A novel heterozygous transversion c.3225T>G, p.(Tyr1075*) in MYO6 gene was identified as the disease-causing variant in this family. This stop-gained variant is predicted to form a truncated myosin VI protein, which is devoid of crucial cargo-binding domain. PCR-RFLP screening in 200 NSHL cases and 200 normal-hearing controls showed the absence of this variant indicating its de novo nature in the population. Furthermore, we reviewed MYO6 variants reported from various populations to date.
Conclusions:
To the best of our knowledge, this is the first family with MYO6-associated hearing loss from an Indian population. The study also highlights the importance of deafness gene panels in molecular diagnosis of GJB2-negative pedigrees, contributing to genetic counseling in the affected families.
Introduction
Hearing loss is a common sensory deficit and has been graded as the third most common cause of years lived with disability in the Global Burden of Disease study (Haile et al., 2021). Etiologically, it can be explained by a multitude of genetic and environmental factors. Around 50% of the cases have an influence of hereditary factors in which 80% of the hereditary hearing loss events are non-syndromic. Nonsyndromic hearing loss (NSHL) is largely a monogenic disease with high genetic heterogeneity, ethnic variability, and multiple inheritance patterns. Variants in more than 100 deafness genes underlie the basis of its pathogenesis (Sheffield and Smith, 2019). To date, 124 genes have been attributed to NSHL with 51 genes implicated in autosomal dominant, 77 in autosomal recessive, and 5 in X-linked hearing loss (Hereditary Hearing Loss Homepage: https://hereditaryhearingloss.org/).
Similar to other world populations, GJB2 is the most investigated gene in India with its pathogenic variations having an overall prevalence of 22.5% amid regional disparity in their frequencies (Yan et al., 2015). Other genes identified such as OTOF, TMPRSS3, TMC1, USH1C, MYO15A, and MYO7A have very less contribution to hearing loss in the Indian population (Yan et al., 2015). Currently, genetic testing rarely influences clinical decision-making, yet genetic studies in hearing loss are typically centered on the prelingual or congenital hearing loss population in India. Individuals with postlingual hearing loss are not frequently tested for genetic etiology in clinical settings with a single cohort study reported to date from India which excluded the involvement of KCNQ variations in postlingual NSHL from Bengali population of India (Adhikary et al., 2017).
Myosin dysfunction has been associated with both prelingual and postlingual forms of hearing loss worldwide. Myosins are motor proteins having a conserved N-terminal motor domain and a variable cargo-targeting region at the C-terminus. Out of 13 different classes of myosins expressed in mammals, pathogenic variants in 6 myosin genes, MYO3A, MYO6, MYO7A, MYO15A, MYH9, and MYH14, have been reported in different forms of human hearing loss (Friedman et al., 2020). Animal models such as mice and zebrafish support the contribution of these myosins in normal hearing (Kappler et al., 2004; Seki et al., 2021).
Myosin VI protein, encoded by MYO6 gene, is the only unconventional myosin that moves toward the minus end of the actin filaments (Wells et al., 1999). Myosin VI is localized to the basal region of inner ear hair cell stereocilia and serves as an anchor, maintaining the structure of the stereocilia (Seki et al., 2021). In humans, MYO6 pathogenic variants are associated with both recessive (DFNB37) and dominant (DFNA22) forms of hearing loss (Ahmed et al., 2003; Melchionda et al., 2001).
In the present study, we identified a novel stop-gained variant in MYO6 gene in a large Indian family affected with postlingual NSHL.
Materials and Methods
Participants
A total of 250 unrelated cases having prelingual NSHL were recruited for the study from ear, nose, and throat (ENT) units of Capital Hospital, Bhubaneswar and SCB Medical College and Hospital, Cuttack, Odisha, India. Among the cases enrolled, a large family with postlingual NSHL was reported (Fig. 1). All the individuals were evaluated via pure-tone audiometry in a double-walled soundproof chamber, according to the standard protocols. The control group consisted of 200 randomly selected individuals from the same ethnic group, having no history of hearing loss among their family members. A written informed consent was obtained from all the participants, and the study was carried out in accordance with the approved guidelines of the institutional ethics committee.

Family affected by postlingual nonsyndromic hearing loss with audiometric profiles and co-segregation of novel MYO6 variant.
DNA extraction and GJB2 sequencing
Peripheral blood (5 mL) was collected from all the participants, and genomic DNA was extracted using the rapid nonenzymatic method (Lahiri and Nurnberger, 1991). Sanger sequencing was performed to exclude the GJB2 (NM_144492) pathogenic variants among the recruited individuals. PCR amplification was conducted using the primers listed in Table 1, followed by ExoSap-IT (Affymetrix Inc., USA) clean-up. Purified PCR products were sequenced using the ABI Prism BigDye Terminator cycle sequencing Ready Reaction kit v. 3.1 (Applied Biosystems, CA, USA) in forward and reverse directions. Sequences were determined on an ABI 3500 Genetic Analyzer (Applied Biosystems) and analyzed using Sequencher® software (Gene Codes Corporation, USA).
Primers Used in the Study
Targeted sequencing and variant analysis
Targeted next-generation sequencing (NGS) was performed with the DNA samples of the proband (III:13) and his affected father (II:6) from the postlingual NSHL family shown in Figure 1 using Human Deafness Panel DA3 (Otogenetics Corporation, GA, USA). Briefly, DNA was enzymatically fragmented and sequencing library was prepared with the DA3 panel targeting 129 deafness genes (Supplementary Table S1). Captured library was sequenced on Illumina platform using 100 bp paired-end reads. The sequencing raw data were subjected to BWA+GATK-Lite (2.3) SNV/indel calling pipeline using human reference genome GRCh37 (hg19) generating variant call format files. All the variants were annotated using wANNOVAR (https://wannovar.wglab.org/) server. A series of filtering criteria were applied to obtain the potentially pathogenic candidate variants from the pool of annotated variants (i) global allele frequency filter (variants absent or reported with a minimum minor allele frequency of 0.005 in public population databases—1000G, ExAC, and gnomAD), (ii) variant class filter (coding and splice site variants excluding synonymous variants), (iii) variants with CADD phred score ≥ 20 or no CADD score, and finally, (iv) heterozygous variants shared among the proband and his father were identified as candidates.
The prioritized candidate variants were classified for their pathogenic or benign nature according to American College of Medical Genetics and Genomics-Association for Molecular Pathology (ACMG-AMP) sequence variant interpretation guidelines and variant interpretation platform for genetic hearing loss (Peng et al., 2021; Richards et al., 2015). The single nucleotide polymorphism database (dbSNP; https://www.ncbi.nlm.nih.gov/snp/) and the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar) were used to check the novelty of the variants and assess the clinical information associated with the variants, respectively. Indian population database, IndiGenomes was utilized to determine the allele frequency of identified candidate variants in the Indian population (Jain et al., 2021).
Validation of candidate variants
Primers were designed flanking the prioritized candidate variants using Primer3 program (https://primer3.ut.ee/) (Table 1). These variants were checked for their segregation with hearing loss in the proband’s family by Sanger sequencing. The identified causal variation in MYO6 gene was screened in controls (n = 200) and GJB2-negative cases (n = 200) by PCR-RFLP method using BstYI and Sanger sequencing. The pathogenicity of the variant was estimated by in silico predictors available on VarSome platform (https://varsome.com/). The conservation analysis of protein sequences between humans and selected species was performed by multiple sequence alignment using Clustal Omega tool (https://www.ebi.ac.uk/Tools/msa/clustalo/). The Swiss-Model (https://swissmodel.expasy.org/) online homology modeling server was used for modeling wild-type and mutant myosin VI protein structures. The modeled structures were viewed and aligned using the PyMOL Molecular Graphics System, Schrödinger, LLC.
Results
Clinical findings in the family
The NSHL family recruited in this study had 10 members affected by postlingual hearing loss. The pedigree study showed autosomal dominant inheritance of hearing loss in this family (Fig. 1A). Affected members of the family had bilateral sensorineural hearing loss (SNHL) of varying intensity (Table 2). Audiograms of the proband (III:13) showed moderate SNHL in the right ear and mild-to-moderate SNHL in the left ear and, his father (II:6) had bilateral moderately severe sloping to profound SNHL in both ears (Fig. 1B, C).
Details of the Family Members Recruited for the Study and Co-segregation of MYO6 Variant
NA, not applicable; SNHL, sensorineural hearing loss.
Identifying the causal variant in the family
Analyzing the GJB2 coding region revealed absence of pathogenic variations in the postlingual NSHL family and 200 prelingual NSHL cases. Targeted NGS analysis for proband and his father produced sequencing reads at mean depth greater than 150X for both the samples. Panel coverage was 99.3% for proband and 96% for father at 20X read depth. Analysis showed 578 and 480 coding region variants in the proband and his father, respectively. Our variant filtration approach detected three heterozygous variations in MYO3A, MYO6, and MYH14, which were common to both proband and his father (Fig. 2, Table 3). Of these three variants, MYO6 c.3225T>G; p.(Tyr1075*) variant was found to be novel (Fig. 3), whereas the other two variants were reported in public databases. Segregation analysis revealed that this novel heterozygous variant co-segregated with hearing loss in an autosomal dominant manner (Fig. 1A, Table 2). This variation was absent in databases — gnomAD (https://gnomad.broadinstitute.org/), IndiGenomes, Deafness Variation Database (https://deafnessvariationdatabase.org/) and our in-house case-control cohort of 400 individuals.
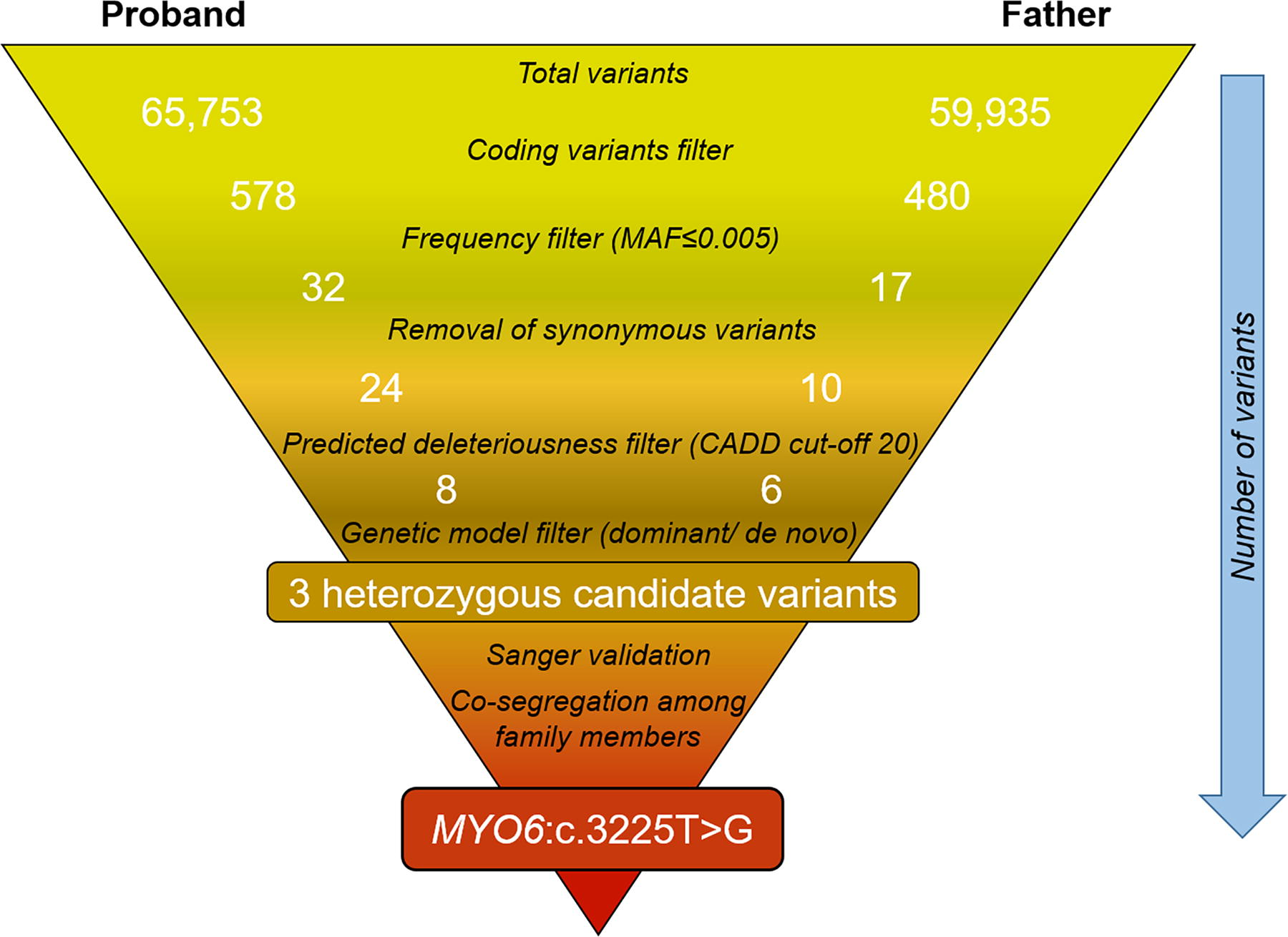
Filtration of variants obtained from targeted NGS analysis of deafness genes in the family with postlingual sensorineural hearing loss. CADD, combined annotation-dependent depletion score; MAF, minor allele frequency; NGS, next-generation sequencing.

Representative electropherograms showing the identified MYO6 variant. The arrows indicate
Genetic Variants Identified in the Indian Family with Postlingual Sensorineural Hearing Loss
Protein multiple sequence alignment of myosin VI among different species showed conservation of 83% of human Tyr1075 residue with a conservative substitution by histidine in some species (Fig. 4A). Homology modeling and subsequent alignment of myosin VI wild-type and p.(Tyr1075*) mutant structures revealed location of the variant in the tail region of protein causing truncation at codon number 1075 leading to deletion of 211 amino acids from myosin VI C-terminal which comprises ubiquitin-binding domain (MyUb) and cargo-binding domain (CBD) of the protein (Fig. 4B, C). Pathogenicity prediction tools—CADD, DANN, and Mutation Taster with scores 36, 0.99, and 0.81, respectively, proposed this variant as disease causing. The deleteriousness meta-score, BayesDel (ranging from −1.293 to 0.757) scored the variant with 0.625 predicting it as pathogenic. According to ACMG-AMP guidelines, this novel variant met the PVS1, PM2, PP1, and PP4 criteria and, hence, could be classified as pathogenic.
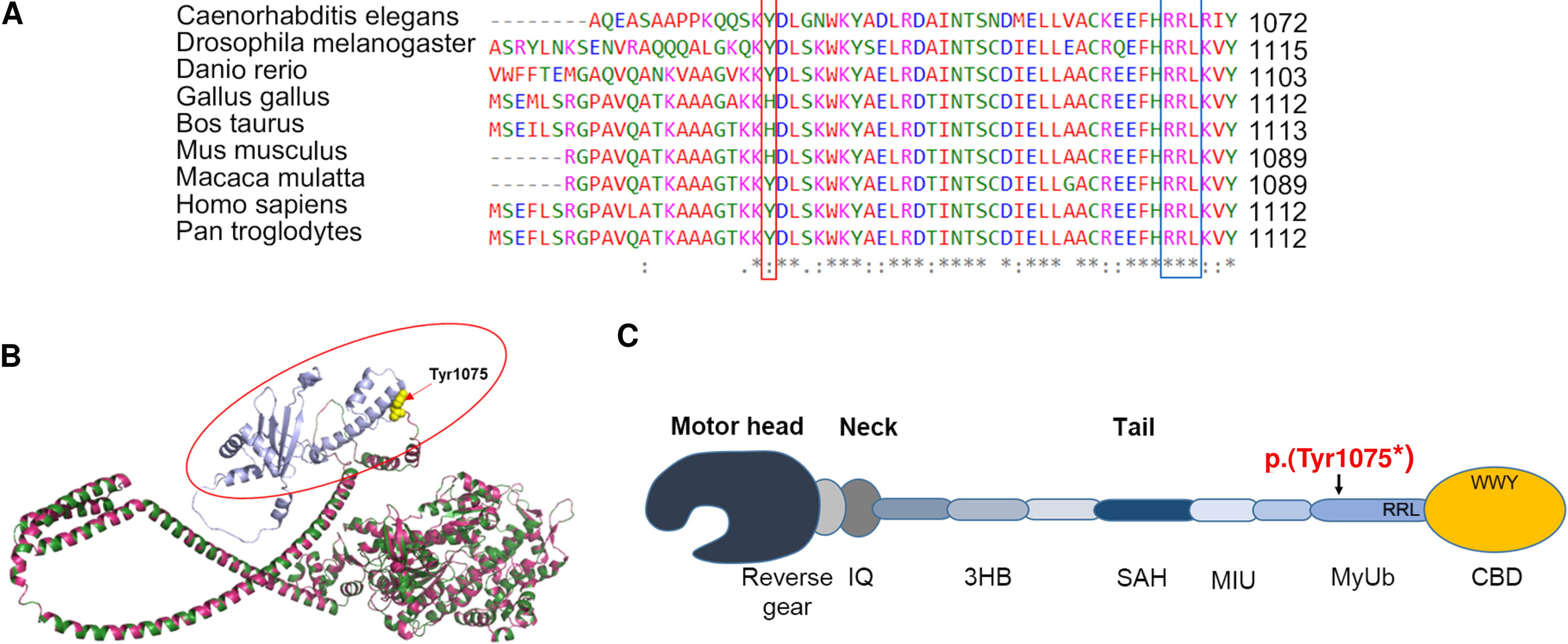
Illustration of MYO6 variant at protein level.
Discussion
Hereditary SNHL is a genetically heterogeneous disorder with diverse clinical forms and modes of inheritance. Postlingual hearing loss majorly inherits in autosomal dominant manner and may have varying audiological profiles (Sheffield and Smith, 2019). Several studies have followed candidate gene and NGS approaches for detecting causal variants in various populations (Vanniya et al., 2022; Yan et al., 2015). In this study, we used a targeted NGS panel of known deafness genes to a multigenerational family with postlingual SNHL and identified a novel heterozygous nonsense variant in MYO6 gene segregating with hearing loss in an autosomal dominant fashion.
Myosin VI expression is reported in inner ear sensory epithelia of mice, zebrafish larvae, and in the human fetal cochlea (Avraham et al., 1995; Scheffer et al., 2015; Seiler et al., 2004). In mice inner ear, myosin VI is localized to inner and outer hair cells particularly concentrated to the cuticular plate and stereocilia rootlets (Avraham et al., 1995) and also synaptic active zone of inner hair cells (Roux et al., 2009). Myosin VI stabilizes stereocilia in hair cells by anchoring the stereocilia to the cuticular plate. MYO6 mutants showed specific characteristics of stereocilial fusion that further led to disorganized and degenerated stereociliary bundles in mice and zebrafish (Seiler et al., 2004; Seki et al., 2017; Self et al., 1999).
Myo6 was first identified as a deafness gene in Snell’s waltzer (sv) mice (Avraham et al., 1995). In humans, MYO6 gene variants have been associated with both autosomal-dominant (DFNA22) and autosomal-recessive (DFNB37) deafness. The first MYO6 variant p.Cys442Tyr in human hearing loss was reported in a large Italian family with postlingual progressive SNHL (Melchionda et al., 2001). This variant was present in the motor domain of myosin VI protein and inherited in the family in an autosomal dominant fashion (DFNA22) and was later proved to be associated with increased ADP dissociation rate possibly hampering the processive movement of myosin VI along actin filaments (Sato et al., 2004). Recently, humanized knock-in mouse model of Myo6 p.Cys442Tyr recapitulated the progressive postlingual sensorineural deafness in humans and showed that the human variant inherits in a semi-dominant pattern (Wang et al., 2019). Homozygous DFNB37 alleles causing recessive deafness were identified later in Pakistani families affected with congenital profound deafness (Ahmed et al., 2003).
The deafness variation database catalogs 19,928 variants in MYO6 gene of which only 32 are nonsense variants, 15 of them are classified as pathogenic, 3 as likely pathogenic, and 14 as variants of uncertain significance. However, there are only 233 hearing loss-associated MYO6 variants reported so far in ClinVar, of which 30 are categorized as pathogenic/likely pathogenic, 59 as having conflicting interpretations of pathogenicity, 108 as variants of uncertain significance, and 36 as benign/likely benign variants. Out of 233 hearing loss-associated variants, only 09 nonsense variations are reported in ClinVar, all of which have been categorized as pathogenic/likely pathogenic variants. Literature mining revealed 63 variants in MYO6 gene reported from different populations across the world, of which most of the variants are associated with autosomal dominant or postlingual forms of hearing loss (Table 4).
MYO6 Variants Identified in the Individuals with Hearing Loss Worldwide
Variant coding region and amino acid coordinates are according to MYO6 transcript NM_004999.
AA, amino acid; AD, autosomal dominant; AR, autosomal recessive.
In Indian population, several genes have been implicated in NSHL and were categorized as second-tier genes for hearing loss in India after GJB2 (Singh et al., 2017; Vanniya et al., 2022; Yan et al., 2015). Most of these studies are primarily concentrated at prelingual NSHL cases, except for a report from West Bengal, India, which ruled out the involvement of KCNQ causal variations in their postlingual NSHL cohort (Adhikary et al., 2017). In the present study, we report the implications of MYO6 gene in NSHL in an Indian population for the first time. This variant was not found in 400 ethnically matched control alleles and Indian population databases, revealing its extremely rare nature.
Previous reports have shown that a single variant can cause both dominant and recessive forms of hearing loss (Table 4). Hence, we speculated that the homozygosity of this novel MYO6 variant could be identified in our congenital NSHL cohort; however, its absence in 200 prelingual cases might be because it originated de novo in the reported family. To understand the contribution of MYO6 causal variants in Indian population, more families should be analyzed by targeted NGS approach or whole exome sequencing. In a recent large-scale genetic analysis in 1,336 Japanese autosomal dominant NSHL families, 2.4% of the families showed 27 variants in the MYO6 gene, among which 22 variations were found to be novel (Table 4) (Oka et al., 2020).
MYO6 pathogenic variants are more frequent in the motor domain, the largest domain of myosin VI, followed by an equivalent number of variants in downstream domains of the protein. The semi-conserved residue p.Tyr1075 lies in the C-terminal tail region of myosin VI upstream of a conserved CBD domain. The transversion, c.3225T>G, generates a premature stop codon leading to protein truncation at myosin VI tail, causing deletion of MyUb and CBD domains of the protein (Fig. 4). Cargo binding at C-terminal tail regulates motor activity of myosin proteins along actin filaments. Loss of CBD restricts myosin VI in a primed nonmotile conformation, affecting intracellular actin filament dynamics which may impede normal Myosin VI function within the hair cells of the inner ear (Arden et al., 2016).
The novel variant is projected to be disease-causing by in silico meta-predictors and meets the pathogenic classification based on ACMG-AMP criteria. Furthermore, this variant is located upstream of a known loss-of-function stop-gained variant c.3496C>T (p.Arg1166*) that causes both recessive and dominant forms of NSHL, possibly because of autosomal semi-dominant inheritance mode (Oka et al., 2020). Previous studies showed a damaging effect of the p.Arg1166* variant resulting in a truncated protein devoid of C-terminal 120 amino acids, which led to aberrant re-localization of myosin VI from intracellular vesicles to actin filaments and abnormally increased actin binding ability (Arden et al., 2016). Presence of this functionally validated, pathogenic nonsense variant (p.Arg1166*), downstream to the novel Indian variant p.(Tyr1075*), further advocates its pathogenicity in NSHL.
The family reported here showed bilateral hearing loss of varying severity, which is in accordance with earlier reports on patients with DFNA22 who developed high-frequency progressive hearing loss possibly owing to loss of outer hair cells because of MYO6 haploinsufficiency (Oka et al., 2020). Recently, researchers achieved CRISPR/Cas system-based RNA base editing in the Myo6 p.Cys442Tyr mouse (Myo6C442Y/+) model, which partially rescued the auditory function in the animal. This supports therapeutic opportunities for MYO6 (DFNA22)-based hearing loss (Xiao et al., 2022). Advancements in NGS-based diagnosis and prospective research in gene editing therapies hold promising possibilities for the prevention of hearing loss in families affected by NSHL.
Conclusions
In this study, we have identified a novel causal MYO6 variant c.3225T>G inheriting in a multigenerational family with postlingual NSHL in an autosomal dominant manner. This is the first report on the implications of MYO6 in hearing loss from Indian population. Limited reports on postlingual hearing loss in India pose an absolute need to address such cases in order to identify causal factors prevailing in the population. This could also help to identify novel targets to prevent NSHL and discover novel mechanisms for comprehending the complex etiology of postlingual hearing loss. Although we are reporting here the first family with MYO6-associated hearing loss from India, further work in this direction may help in determining the contribution of MYO6 genetic variants in NSHL in India. The study also highlights the significance of deafness gene panels in determining the underlying genetic factors prevalent in the Indian population, which is important in prenatal diagnosis and genetic counseling in the affected families.
Footnotes
Acknowledgments
The authors thank all the individuals who participated in this study.
Authors’ Contributions
Conceptualization: P.V.R. Methodology: R.R. and P.V.R. Formal analysis and investigation: R.R., P.V.R., K.C.P., and C.S.R. Writing—original draft preparation: R.R. and P.V.R. Writing—review and editing: P.V.R. and R.R. Resources: P.V.R., K.C.P., and C.S.R. All authors read and approved the final article.
Data Availability
The research data included in this article can be obtained from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
This study was approved by the Institutional Ethical Committee of Institute of Life Sciences, Bhubaneswar and SCB Medical College and Hospital, Cuttack, India, and written informed consent was obtained from all participants.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Intramural funding from the Institute of Life Sciences (ILS), Bhubaneswar. The ILS is funded by the Department of Biotechnology, Govt. of India.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.