
Abstract

Introduction
VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome is an adult-onset severe systemic autoinflammatory monogenic syndrome with progressive hematological manifestations. The syndrome was described in 2020 by Beck et al. who discovered the disease in 25 men with severe, late-adult-onset autoinflammatory disease (Beck et al., 2020). The disease is important because it can be a source of severe morbidity and mortality, which was not previously recognized, and the patients can potentially benefit from specific therapies once recognized. It is also a prototype for a new class of “hematoinflammatory diseases” that shows prominent and diverse hematological and autoinflammatory symptoms (Grayson et al., 2021). Since its discovery, many more cases have been reported, with the true frequency of this condition still to be determined, with evidence that the disease remains an underdiagnosed entity (Beck et al., 2023; Georgin-Lavialle et al., 2022).
Clinical Manifestations
VEXAS syndrome is a heterogeneous disease that can present with diverse hematological and inflammatory manifestations. The most common clinical features of VEXAS syndrome comprise skin lesions, pulmonary involvement, arthralgia/arthritis, ear/nose chondritis, thromboembolic disease, recurrent fever, cytopenias, and various types of vasculitis (Uchino et al., 2022) (Fig. 1). Rheumatologic features often overlap with hematological disorders, making diagnosis challenging. Many patients may present with autoinflammatory diseases such as relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, and/or myelodysplastic syndrome (MDS). Bone marrow evaluation is generally indicated to exclude other potential causes of cytopenias and/or associated plasma cell neoplasms. The gold standard for diagnosis is genetic confirmation of ubiquitin-activating enzyme 1 (UBA1) mutations (Loeza-Uribe et al., 2024), which can be identified on peripheral blood, bone marrow, or involved tissue specimens such as those involved by Sweet syndrome (Gurnari et al., 2022). VEXAS is associated with high morbidity and mortality (Vergneault et al., 2023) due to hematological manifestations including cytopenias, macrocytic anemia, MDS, monoclonal gammopathies, and poorly controlled severe inflammatory symptoms, which are often resistant to treatment (Beck et al., 2023).
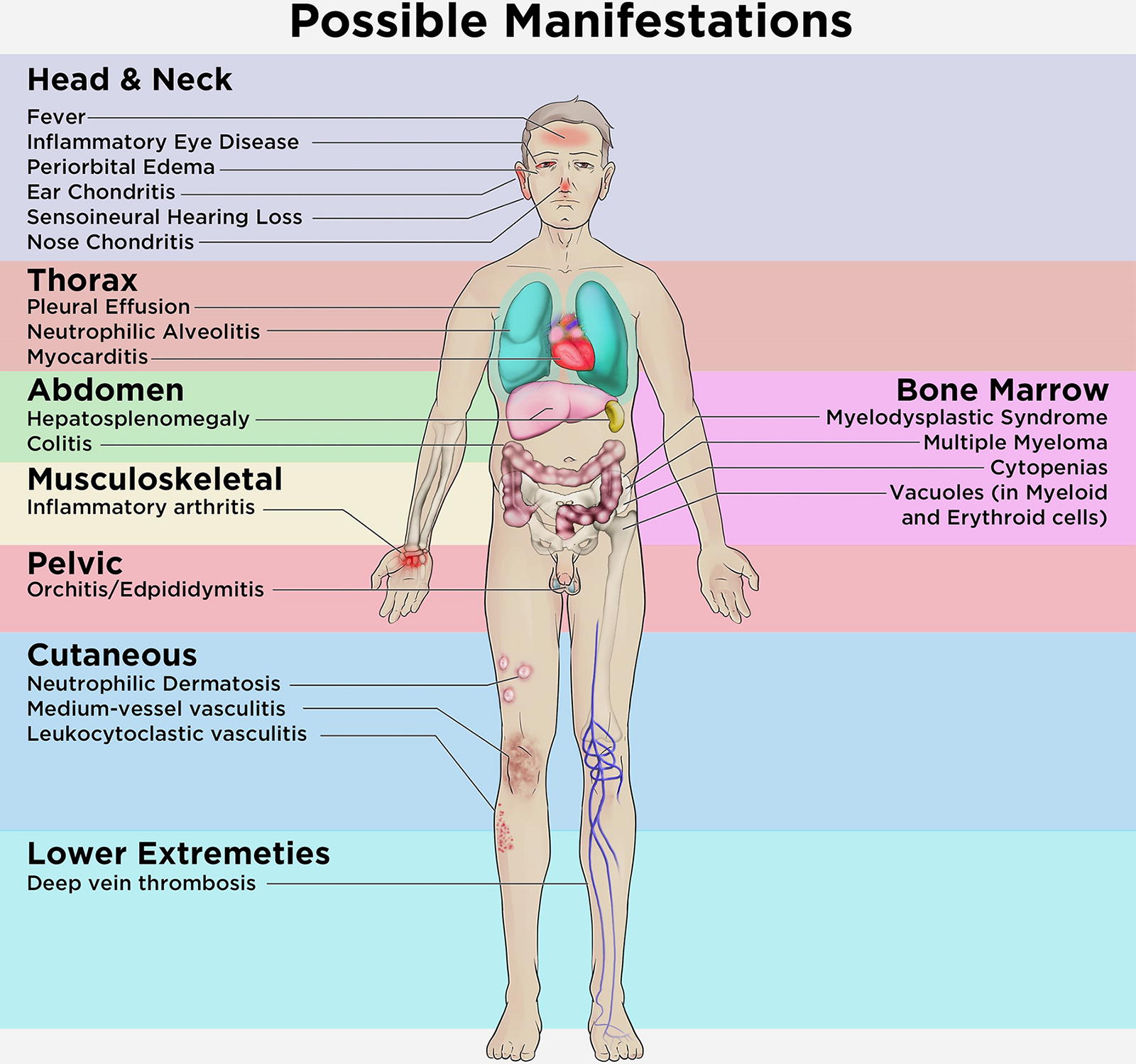
VEXAS symptoms compared to all the possible manifestations (adapted from [Grayson et al., 2021]).
Currently, there are no established treatment guidelines for VEXAS syndrome; however, there are multiple potential therapies emerging that have shown efficacy in at least a subset of patients. The use of azacytidine and/or ruxolitinib seems promising; however, larger clinical trials to confirm these findings are recommended (Raaijmakers et al., 2021; Heiblig et al., 2022). In general, the management of patients relies on targeting immune and inflammatory pathways or targeting the UBA1-mutated hematopoietic population (Khitri et al., 2024), based on experience gained from the treatment of other autoinflammatory diseases or of reported VEXAS cases. Some patients may have a marked response to therapy, while others may show resolution of some symptoms, especially skin, rheumatic, and/or hematological manifestations. Moreover, many patients become unresponsive even after several different treatments, thus increasing the mortality rates (van der Made et al., 2022). Stem cell transplantation may be considered in severe cases and is potentially curative (Ali and Gurnari, 2024). Overall, more research, especially large multicentric randomized clinical trials, will be crucial in creating more robust management protocols.
Genetics of VEXAS syndrome
VEXAS is caused by acquired somatic mutations in the UBA1 gene (Beck et al., 2023). UBA1 encodes the main E1 activating enzyme in humans, which is responsible for ubiquitin activation, ubiquitylation-dependent intracellular protein degradation, and cell homeostasis (Moudry et al., 2012). Beck et al. estimated the prevalence of UBA1 mutations to be 1:4269 (95% CI: 1:2319-1:7859) among men and 1:26,238 (95% CI: 1:7196-1:147,669) among women aged over 50 years with an overall prevalence of 1:13,591 (95% CI: 1:7775-1:23,758) and a disease penetrance of 100% (Beck et al., 2023).
VEXAS syndrome is an X-linked disease that predominantly affects men as well as women with X chromosome monosomy (Stubbins et al., 2022); generally women show milder clinical manifestations, possibly because the additional unaffected allele protects against the effects of the mutant allele in these patients (Beck et al., 2023). The UBA1 mutations described originally in VEXAS syndrome were all missense in methionine 41 (Met41) (Beck et al., 2020). Mutations in p.Met41 were later confirmed to be the most common mutation in VEXAS syndrome patients (Gutierrez-Rodrigues et al., 2023). These mutations disrupt the synthesis of the cytoplasmic isoform, UBA1b, and instead lead to the expression of UBA1c (also referred to as UBA1mut), an isoform with reduced catalytic activity that leads to endoplasmic reticulum stress in the myeloid cells; in response, these cells express higher levels of inflammatory cytokines, thereby driving inflammation (Bourbon et al., 2021; Beck et al., 2020; Gutierrez-Rodrigues et al., 2023). UBA1 mutations likely trigger the activation of inflammatory pathways, resulting in severe systemic inflammatory symptoms (Poulter et al., 2021).
The most common mutation in VEXAS syndrome is the missense mutation p.Met41Thr. Other common mutations include p.Met41Val, p.Met41Leu, and p.Ser56Phe (Gutierrez-Rodrigues et al., 2023; Poulter et al., 2021; Bourbon et al., 2021) (Table 1). Met41 substitutions are primarily found in myeloid lineage cells, including bone marrow hematopoietic progenitor cells and circulating myeloid cells. Few cases show mutations affecting the splice acceptor site immediately before exon 3, possibly causing loss of Met41, as this is the second amino acid encoded by exon 3. Hence, these splice site mutations also lead to UBA1c expression. Apart from these common mutations, other mutations in UBA1, not involving Met41, have been reported. (Table 1) A new mutation in the splice motif at the junction of intron 2 and exon 3 (c.118-2A>C and c.119-1G>C) was found in two patients, which resulted in the loss of four amino acids in the UBA1 protein, including Met41 (Bourbon et al., 2021). Two additional variants, each in one patient, were identified: the first variant (C.167C>T; p.Ser56Phe) was restricted to myeloid lineage and led to temperature-dependent impairment of UBA1 catalytic activity; the second, located at the splice acceptor site (NM_153280:c.118-1G>C) of exon 3, led to fewer properly spliced transcripts and defective incorrectly spliced products (Poulter et al., 2021). Six novel somatic mutations in UBA1 leading to VEXAS syndrome were also reported (p.His55Tyr, p.Gly477Ala, p.Ala478Ser, p.Asp506Gly, p.Asp506Asn, and p.Ser621Cys), with none causing UBA1c production but instead leading to reduced catalytic activity of UBA1a and UBA1b (Collins et al., 2022). Atypical form of VEXAS syndrome has been reported due to mutations in the active adenylation domain (AAD) of UBA1, which is essential for ubiquitin activation and targeted by mutations in inherited X-linked spinal muscular atrophy (Faurel et al., 2023). UBA1 AAD mutant patients expand the spectrum of diseases associated with clonal hematopoiesis with somatic UBA1. As compared with typical VEXAS patients, UBA1 AAD patients have less autoinflammatory manifestations, except of skin lesions and joint disease, but notable marked erythroid hyperplasia (Faurel et al., 2023).
List of Genetic Mutations Common in VEXAS Syndrome
It remains unclear whether VEXAS arises due to UBA1b loss or UBA1c appearance. However, disease manifestation and severity may rely on the type of Met41 substitution and the resulting balance between UBA1b/UBA1c proteins. Specifically, among all possible methionine substitutions, only those to threonine, valine, and leucine have been associated with disease, suggesting that these specific amino acid changes may allow sufficient protein functionality to support cell viability while still contributing to pathogenesis. In contrast, other substitutions may result in more profound disruptions of UBA1 function, potentially rendering cells nonviable. For instance, the Met41Val is associated with worse prognosis (Ferrada et al., 2021), potentially due to more severe functional impairment of UBA1, while Met41Leu appears to be associated with better outcomes (Georgin-Lavialle et al., 2022), possibly due to a lesser degree of functional disruption. These observations raise the possibility that the functional and structural compatibility of the resulting protein plays a crucial role in determining both disease presence and severity. Further studies are required to systematically evaluate the effects of alternative Met41 substitutions on protein stability, enzyme activity, and cellular survival to better understand these associations. These results highlight how varying mutations in the same amino acid can drive different phenotypes and may provide insight for understanding VEXAS pathogenesis and that of other autoinflammatory disorders. Recent research has also highlighted the importance of genetic testing for UBA1 mutations in patients with nonspecific inflammatory symptoms, and this led to a diagnosis in a subset of these patients (Beck et al., 2023). Further research is warranted to decipher the underlying mechanisms and best practice guidelines for pursuing testing.
Hematological manifestations and hematological malignancies
VEXAS syndrome patients present with hematological manifestations and increased predisposition to hematological malignancies (Gutierrez-Rodrigues et al., 2023). The most common hematological manifestation is macrocytic anemia and is found in up to 96% of the patients (Beck et al., 2020). Other manifestations include progressive bone marrow failure, thrombocytopenia, and thromboembolic disease (Grayson et al., 2021). Thrombotic complications are reported to be around 40% (Beck et al., 2020), of whom most present with venous thromboembolic disorders rather than arterial (Oo et al., 2022). An increased risk for hematological malignancies, such as MDS and plasma cell neoplasms, has been reported in VEXAS syndrome patients (Gutierrez-Rodrigues et al., 2023). Dysplasia in the form of vacuoles in bone marrow myeloid and erythroid precursor cells is a characteristic of the disease (Obiorah et al., 2021; D’Angelo, 2023). However, these vacuoles are nonspecific, not present in every case, and may be present in only a small fraction of cells. (Lacombe et al., 2024). The presence of vacuoles in myeloid cells should always warrant further investigations since the differential is wide and includes alcohol intoxication, copper deficiency/zinc toxicity, and myeloid neoplasms (Grayson et al., 2021). The exact content of the vacuoles in VEXAS patients remains unclear and warrants further investigation and research (Grayson et al., 2021). It is important to note that the hematological manifestations in VEXAS patients are usually significant; however, only a subset of cases have enough evidence to be diagnosed as MDS (Obiorah et al., 2021; Gutierrez-Rodrigues et al., 2023).
Patients with VEXAS syndrome have a higher frequency (60%) of typical clonal hematopoiesis (CH) mutations, particularly in DNMT3A and TET2 (Gutierrez-Rodrigues et al., 2023); however, these genes were not found to affect phenotypic heterogeneity or disease severity (Gutierrez-Rodrigues et al., 2023). Interestingly, the VAF of UBA1 mutations observed was usually high (median: 74%) (Gutierrez-Rodrigues et al., 2023). While the presence of typical CH mutations in VEXAS syndrome is interesting, these genes are not specific to VEXAS and are present across many other immunological diseases. Most of the VEXAS-related MDS cases showed few mutations and a normal karyotype. The vast majority of VEXAS patients with MDS were classified as low-risk MDS as shown in a large case series (Gutierrez-Rodrigues et al., 2023). Most of these cases did not progress to higher grade MDS/AML, with recent research recommending creating a distinct entity, UBA1 mutated MDS, to differentiate the disease from other higher risk MDS entities by standard criteria (Gutierrez-Rodrigues et al., 2023). Overall, the diagnosis of MDS in the setting of VEXAS is challenging, and many patients had an aggressive course despite apparently low-risk disease by standard criteria. These observations support the proposal that MDS/VEXAS cases should be closely followed until enough evidence accumulates to possibly create a separate category for these cases.
While MDS has been shown to be associated with VEXAS syndrome patients (Gutierrez-Rodrigues et al., 2023), MDS have also been reported across many other rheumatologic diseases (Mobini et al., 2015). Patients with VEXAS have a distinct high propensity for developing MDS, with the frequency of the condition ranging between 20% and 55% (Beck et al., 2020; Poulter et al., 2021; Bourbon et al., 2021; Georgin-Lavialle et al., 2022). While up to 10% of MDS patients have no established disease-defining mutations based on the myeloid next-generation sequencing panels, the addition of UBA1 mutations could certainly decrease this percentage (Sirenko et al., 2024). A causative link between UBA1 mutations and myeloid neoplasm or the occurrence of MDS in VEXAS remains to be established by future studies. VEXAS syndrome is certainly an addition to the growing list of hematoinflammatory diseases spectrum. STAT3 mutations have been linked to Felty syndrome and Large granular lymphocytic leukemia (LGL); BRAF mutations have been linked to Erdheim-Chester disease and aortitis; Sjogren syndrome has been found to be associated with B-lymphomas such as marginal zone lymphoma (Grayson et al., 2021).
Conclusion and Future Directions
VEXAS syndrome is a recently defined disease falling within the category of “hematoinflammatory diseases.” VEXAS syndrome may serve as a prototype of these disorders; however, several unknown aspects concerning the disease still exist. These include the role of additional mutations in disease progression, risk stratification, the full genetic mutational spectrum of the disease, and the optimal management/surveillance approach for these patients. The disease should be investigated in cases presenting with treatment-refractory inflammatory disease and progressive hematological abnormalities. Early research showed that the disease is possibly caused by just few a mutations; however, the last few years showed that the disease genetics is more complex than what was initially thought and that the spectrum of genetic mutations can be wide (Table 1). Observations in VEXAS syndrome and related diseases can help define the role of acquired somatic mutations. The overlap of VEXAS clinical manifestations with those of other inflammatory conditions, as well as its high morbidity and mortality, warrants a multidisciplinary approach for accurate diagnosis and optimal management of these patients; unfortunately, many of these patients still remain undiagnosed (Beck et al., 2023). Setting distinct guidelines for diagnosis and management should be a high priority in the coming few years (Beck et al., 2023). The recently developed international AutoInflammatory Disease Alliance registry is expected to aid in this direction (Vitale et al., 2022). Reporting of new cases remains important to gain insight into the heterogeneous and complex nature of the disease. Some significant issues that seem to hinder our understanding of the disease include the lack of a higher animal model, limited provider/public knowledge about the disease, and the limited availability of the gene in commercial panels (Grayson et al., 2021). Currently, UBA1 gene mutation assessment is still not available in most standard workup panels for myeloid neoplasms or immune dysregulation panels. The possibility of lack of insurance coverage for the patients and prior insurance pre-authorization represents another significant hurdle. With the discovery of VEXAS and the ability to perform broader genetic panels at lower costs, we believe that diagnosis of these patients will improve. Further, as the knowledge of this disorder improves, the identification of these cases will also increase.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.