
Abstract
Background:
Aplasia cutis congenita and ectrodactyly skeletal syndrome (ACCES) is caused by heterozygous variants in the UBA2 gene, with phenotypic heterogeneity encompassing a range of diverse skeletal, dermatological, and neurological features.
Aims:
The goal of our research was to suggest that pathogenic frameshift variant c.52_58dupGGCCGGG p.(Val20Gfs*31) could lead to the development of ACCES and also to review the literature to document phenotypic variability among individuals with UBA2 variants, providing further insights into this ultrarare syndrome.
Methods and Result:
We report a case of a 7-year-old male presenting with cutis aplasia congenita, syndactyly, preaxial polydactyly, and severe hypospadias. Exome sequencing (ES) identified a heterozygous frameshift variant c.52_58dupGGCCGGG p.(Val20Gfs*31) in the UBA2 gene. This variant is absent in gnomAD and is predicted to cause a premature stop codon with consequent protein truncation and/or nonsense-mediated decay. Initially classified as a variant of uncertain significance, this frameshift variant was reclassified as pathogenic following a comprehensive reassessment post-enrollment of the patient in the Undiagnosed Rare Disease Clinic of Indiana University School of Medicine.
Conclusion:
This study illustrates the critical role of ongoing genomic data reevaluation, particularly in unsolved cases, where variant reclassification has the potential to impact diagnostic precision, targeted treatment planning, and family counseling. The clinical variability observed among reported cases, spanning mild to severe presentations, underscores the complexity of UBA2-related disorders. This variability suggests an interplay of genetic modifiers, epigenetic influences, and environmental factors, highlighting the need for further research into the mechanisms driving this heterogeneity.
Introduction
The UBA2 gene (MIM:613295) is located at 19q13.11 and encodes a ubiquitin-like modifier activating enzyme. that the enzyme is involved in sumoylation, which is a posttranslational modification that adds small ubiquitin-like modifier (SUMO) proteins to a target protein to change function, structure, or intracellular localization. Although sumoylation can target proteins for degradation, it is also involved in cell cycle regulation, subcellular trafficking, signal transduction, stress responses, chromatin structure dynamics, and molecular mechanisms of disease. In addition, UBA2 forms a heterodimer with SUMO-Activating Enzyme Subunit 1 (SAE1) and binds with SUMO1 in an ATP-dependent manner (Desterro et al., 1999; Yang et al., 2017), which makes an enzyme for the sumoylation of proteins. UBA2 lies adjacent to the minimum overlapping region of the 19q13.11 deletion syndrome. The features of this deletion syndrome include early growth deficiencies, developmental delay, distinctive facial features, aplasia cutis congenita (ACC), hip dysplasia, digital and limb anomalies including ectrodactyly, and other malformations. Loss of function (LoF) of UBA2 has been proposed to underlie key aspects of the deletion phenotype, including ACC and ectrodactyly (Gana et al., 2012; Malan et al., 2009). De novo frameshift, missense, and nonsense variants in UBA2 have been reported in several patients with a range of physical and the development abnormalities. Some of these anomalies include ACC, ectrodactyly, hand/foot malformations, neurodevelopmental disorders, hypotonia, and renal and genital abnormalities (Marble et al., 2017; Parveen et al., 2023; Wang et al., 2020).
We present the case of a 7-year-old male with mild frontal bossing, wide-set eyes (telecanthus), an underdeveloped nasal bridge, a sacral dimple, ACC, syndactyly, pre-axial polydactyly, and severe hypospadias. Initially, the frameshift variant c.52_58dupGGCCGGG p.(Val20Gfs*31) in the UBA2 gene that was identified through ES by a commercial clinical lab (GeneDx) was classified as a variant of uncertain significance (VUS). Subsequently, the patient was enrolled in the Undiagnosed Rare Disease Clinic (URDC), where a detailed review of medical literature and available evidence led to the reclassification of this variant as pathogenic, in accordance with American College of Medical Genetics (ACMG) guidelines. In addition, we reviewed published literature describing 26 individuals from 15 unrelated families with both de novo and familial UBA2 sequence variants. These individuals exhibit highly variable phenotypes with overlapping clinical features, further illustrating the phenotypic spectrum of UBA2-related conditions.
Clinical Presentation and Results
The patient is a 7-year-old adopted male who presented to the URDC due to his complex medical history of ACC, syndactyly, preaxial polydactyly, and severe complex hypospadias. Additional clinical features include mild frontal bossing, telecanthus, hypoplastic nasal bridge, a sacral dimple, and cutis marmorata (Fig. 1A-D). The cutis aplasia measured approximately 1 cm in diameter and was located at the crown of the patient’s head. He exhibits normal development, cognition, and vision. He had an echocardiogram, which showed normal cardiac anatomy and function. Lumbar spine MRI and renal ultrasound were also normal. The parents reported no other health concerns. Biochemical testing was performed and was essentially normal. This testing included free and total carnitine analysis, acylcarnitine profile, plasma amino acid profile, plasma methylmalonic acid, urine organic acid screen, and urine amino acid screen.

Clinical features of a patient carrying UBA2 variant (c.52_58dupGGCCGGG; p.[Val20Glyfs*31]) showing
Genetic testing prior to enrollment in the URDC included chromosomal microarray (CMA) and ES. The CMA was normal, and ES revealed two VUSs, including a frameshift variant c.52_58dupGGCCGGG p.(Val20Glyfs*31) and a missense c.464T>A p.(Val155Glu) in the UBA2 gene. The frameshift variant is absent in the gnomAD database, predicted to cause a frameshift starting with amino acid valine 20 which changes this amino acid to a glycine residue, and creates a premature stop codon at position 31 of the new reading frame. This variant is predicted to cause loss of normal protein function through protein truncation or nonsense-mediated mRNA decay. UBA2 has a high pLI (1.0) score, indicating a strong intolerance to LoF variants (Lek et al., 2016).
However, at the time of the initial clinical ES, only a few cases with de novo missense changes in this gene had been reported. Consequently, due to the lack of parental information to determine inheritance, limited understanding of the molecular mechanism underlying the disorder, and insufficient literature on UBA2-related etiology, this LoF variant was initially classified as a VUS by GeneDx. Following the proband’s enrollment in the URDC, a detailed review of the medical literature revealed new details, including novel genotype-phenotype correlations. Based on this emerging information, the variant was reclassified as pathogenic (ClinVar Accession: VCV000524055.4).
In addition, the second variant, c.464T>A p.(Val155Glu), is a nonconserved amino acid substitution, which is likely to impact secondary protein structure. In silico analyses, including protein predictors and evolutionary conservation, support a deleterious effect. However, it is unknown if the two variants are on the same UBA2 allele (in-cis) or on opposite alleles (in-trans), as the biological parents of the proband did not participate in the study. This variant has been classified as a VUS following the ACMG guidelines.
Discussion
We presented a case of a 7-year-old boy with a complex medical history of ACC, syndactyly, pre-axial polydactyly, and severe hypospadias, all due to a frameshift pathogenic variant c.52_58dupGGCCGGG: p.(Val20Glyfs*31) in the UBA2 gene. UBA2 is composed of 17 coding exons and encodes a protein of 640 amino acids. To date, only seven reports have been published that describe the intragenic pathogenic variants in the UBA2 gene. So far, a total of 26 patients from 15 unrelated families have been identified with 15 pathogenic variants in the UBA2 gene (Fig. 2).
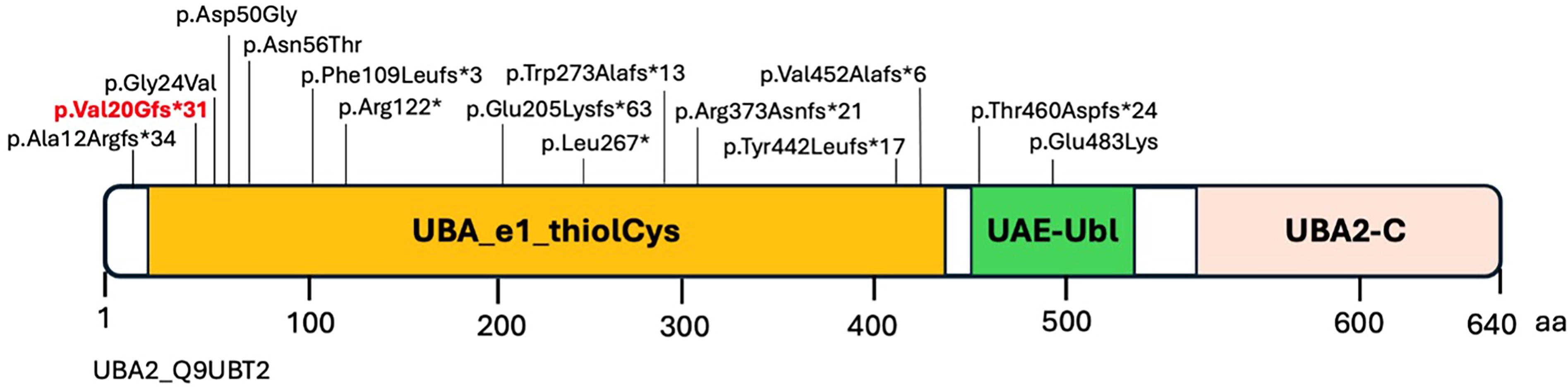
Schematic representation of the UBA2 domains. Shown in red is the novel variant identified in this study; the other variants were previously identified for UBA2-related disorders.
UBA2 has three domains: UBA-e1-thiolCys, UBA-UBL, and UBA2-C. The UBA2 protein domain (UBA-e1-thiolCys) with catalytically active sites of ubiquitin-activating enzymes is shown in yellow (Fig. 2). This domain has putative active sites to bind ATP, substrate, and zinc, with the last of five conserved cysteine residues playing an important role in ubiquitin thioester complex formation. Thirteen out of 15 variants, including the novel variant p.(Val20Glyfs*31) identified in this study, are in the most important UBA-e1-thiolCys domain of UBA2. The UBA2-C and UAE-UbL are C-terminus and ubiquitin-like domains of UBA2 (Fig. 2; Table 1). It has been observed that most of the amino acid residues in the UBA-e1-thiolCys domain are involved in the ATP binding, and substitutions of amino residues in this domain effect the ATP binding and ectopic interactions of amino acids with nearby residues (Lois and Lima, 2005; Schnur et al., 2021).
Overview of the Patients Identified with UBA2 Intragenic Variants
This table has been modified from a previous study9, we have updated this table in the present study by adding the information of additional six patients (S. No. 1 to 6).
UBA2 as a Candidate Gene for Ectrodactyly
De novo frameshift, missense, and nonsense variants in UBA2 have been reported in patients with a range of physical and developmental abnormalities (Marble et al., 2017; Schnur et al., 2021). Previously, it was hypothesized that UBA2 plays a role in the scalp defects in the 19q13.11 deletion syndrome and was suggested as a novel candidate for Mendelian scalp defects and other anomalies (Marble et al., 2017; Melo et al., 2015; Schnur et al., 2021). These anomalies include ACC, ectrodactyly, hand/foot malformations, neurodevelopmental disorders, hypotonia, and renal and genital abnormalities (Marble and Pridjian, 2002; Schnur et al., 2021; Yamoto et al., 2019). Several studies have also identified UBA2 variants associated with split-hand disorders. Recently, pathogenic variants in the UBA2 gene have been identified to cause ectrodactyly with variable expressivity and reduced penetrance in some cases. Two affected sisters, who had features of ectrodactyly, inherited a frameshifting variant c.1118delG p.(Arg373fs) in UBA2 from a phenotypically normal mother. Similarly, reduced penetrance was also observed in a patient carrying a de novo frameshift variant c.612delA p.(Glu205Lysfs*63) in UBA2 (Aerden et al., 2020; Elsner et al., 2021; Parveen et al., 2023). In 2019, UBA2 was identified as a candidate gene for split-hand/foot malformation (SHFM). This role of UBA2 was further confirmed by other studies in both human subjects and animal models (zebrafish) (Aerden et al., 2020; Elsner et al., 2021; Parveen et al., 2023; Yamoto et al., 2019). The characterization of the knockout phenotype in zebrafish supports the significance of UBA2/uba2 in development, potentially by affecting posttranslational modification of SHFM-associated genes (Schnur et al., 2021).
Phenotypic Heterogeneity in UBA2-Related Syndromes
There is high intrafamilial and interfamilial phenotypic heterogeneity among the patients carrying UBA2 variants. The variability lies in the expression of dermatologic, skeletal, neurological, cardiac, and renal features, similar to those of the chromosome 19q13.11 microdeletion syndrome (Aerden et al., 2020; Elsner et al., 2021; Marble et al., 2017; Parveen et al., 2023; Schnur et al., 2021; Wang et al., 2020; Yamoto et al., 2019). Phenotypically, UBA2-related ACCES are characterized by highly variable expressivity, even within the same family. Most patients exhibit scalp defects, while ectrodactyly is less common. However, more variable and less obvious digital and skeletal anomalies are often present. The intrafamilial phenotypic heterogeneity was observed in a family in which a UBA2 frameshift variant c.327delT p.(Phe109Leufs*3) in the proband was inherited from the affected mother. The mother had ACC but was otherwise healthy. The son had ACC, microcephaly, bilateral ectrodactyly, low‐lying conus medullaris, horseshoe kidney, and tracheoesophageal fist (Wang et al., 2020). In 2021, the phenotypes of the patients who possessed intragenic UBA2 variants were compared, and it has been observed that the most specific aspect of UBA2-related phenotypes was ACC, while ectrodactyly was less common (Table 1) (Schnur et al., 2021). Interestingly and subsequently, five patients with intragenic UBA2 variants and limb malformation have been reported. None of these patients had features of ACC (Elsner et al., 2021; Parveen et al., 2023). In this review, we have compared the clinical features of the UBA2-related patients and updated the clinical summary table (Table 1) that was previously described (Schnur et al., 2021). Since a wide phenotypic variability is observed in both human and zebrafish UBA2/uba2-related phenotypes, additional studies are warranted to define potential modifiers (Schnur et al., 2021).
Conclusion
In summary, we report a novel pathogenic loss-of-function variant, c.52_58dupGGCCGGG p.(Val20Glyfs*31), in the UBA2 gene. A thorough literature review, medical record review, exam, and reanalysis of our patient’s UBA2 variants ultimately led to the reclassification of the frameshift variant from VUS to pathogenic. This underscores the critical importance of periodic reassessment of genetic variants through comprehensive literature reviews, database updates, and detailed phenotypic reevaluations. Such integrative approaches provided definitive evidence supporting the pathogenicity of this frameshifting UBA2 variant, bringing a resolution to this family’s diagnostic journey. Addressing the VUS reclassification is essential to enhance the credibility of genetic testing and clinical impact. As genomic databases, such as gnomAD, ClinVar, and Decipher, among others, grow and knowledge advances, we would expect that the reclassification rates would greatly increase, reducing variant classification discordance in the future.
Moreover, our study highlights the strikingly variable expressivity observed in human UBA2-related phenotypes, both within and across affected families. A comparison of clinical features among previously reported UBA2 cases reveals a spectrum of phenotypes, ranging from mild developmental delays to more severe systemic manifestations, and further attests to this variability.
The mechanisms underlying the UBA2 phenotypic heterogeneity remain incompletely understood but likely involve genetic modifiers, environmental influences, or epigenetic factors. Emerging evidence suggests that interacting genes involved in ubiquitination, SUMOylation, or related pathways may act as potential modifiers, influencing the expressivity and severity of UBA2-associated conditions. Identifying such modifier genes and their impact on the UBA2 variant’s pathogenicity could provide deeper insights into the biology of this disorder and refine our understanding of its clinical spectrum.
Ultimately, this case highlights the challenges faced by families during the diagnostic odyssey of ultrarare syndromes and reinforces the importance of integrating genomic reanalysis with detailed phenotypic characterization. Regularly revisiting variant classifications, combined with insights from a growing body of literature, remains essential for improving diagnostic outcomes and advancing personalized patient care.
Footnotes
Acknowledgments
The authors extend their deep appreciation to the two sisters and their family for participating in this study. Thank you to Lili Mantcheva for assisting with IRB requirements, consenting, and communicating with the family during the length of the project.
Authors’ Contributions
F.V. and E.C. proposed the meaning and concept of the study and designed the plan for the case and edited the article. K.T. and E.C. performed the initial medical record review and clinical variant assessment. They collected and analyzed the patient’s clinical and phenotypic data. E.C., M.M., and D.D.W. performed exams. E.C. oversaw the project and assessed all of the data to determine the necessity of additional phenotypic or clinical testing, and contributed to and analyzed these data. F.V. analyzed the data. K.L. and K.F. drafted the article. K.T., M.M., and D.D.W. recruited the family, made contributions to data collection and analysis, and was involved article editing.
Data Availability Statement
Ethics Statement
The research project used medical health information and specimens that had been collected as part of an ongoing research study at the Undiagnosed Rare Disease Clinic at Indiana University. Written informed consent was obtained from participants and/or their legal guardians for the collection, research use, and storage of the specimens according to the protocol approved on July 1, 2020, by the Indiana University Institutional Review Board (IRB# 2005902680).
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study is funded in part by the Indiana University Grand Challenge Precision Health Initiative.