
Abstract
Abstract
Wills, Christopher. Rapid recent human evolution and the acccumulation of balanced genetic polymorphisms. High Alt. Med. Biol. 12:149–155, 20
The first part of this review examines recent studies that have had some success in dissecting out the role of natural selection, especially in humans and Drosophila. Among many examples, these studies include those that have followed the rapid evolution of traits that may permit adaptation to high altitude in Tibetan and Andean populations. In some cases, directional selection has been so strong that it may have swept alleles close to fixation in the span of a few thousand years, a rapidity of change that is also sometimes encountered in other organisms.
The second part of the review summarizes data showing that remarkably few alleles have been carried completely to fixation during our recent evolution. Some of the alleles that have not reached fixation may be approaching new internal equilibria, which would indicate polymorphisms that are maintained by balancing selection. Finally, the review briefly examines why genetic polymorphisms, particularly those that are maintained by negative frequency dependence, are likely to have played an important role in the evolution of our species. A method is suggested for measuring the contribution of these polymorphisms to our gene pool. Such polymorphisms may add to the ability of our species to adapt to our increasingly complex and challenging environment.
Introduction
Second, rapid population expansion is more likely to be a strong indicator of accompanying selection than of the slackening of selective pressures that Jones (1994) has suggested. The most rapid human population expansion for which we have evidence is the dramatic increase in the Mongol Y chromosome in central Asia over the last thousand years, which mirrors the growth of the Mongol empire and has produced a large change in the central Asian and northern Chinese gene pools (Zerjal, 2003). Similar expansions can be traced to selective advantages in other human groups (Xue et al., 2005), including the invention and spread of agriculture that displaced hunter–gatherer groups (Berniell-Lee et al., 2009; Richards, 2003).
We are on the cusp of understanding the rapid evolution produced by our recent cultural and technological changes, along with other genetic changes that took place in the more distant past, in unprecedented detail. This is because we can now follow in detail recent changes that can be detected by sequencing of entire human genomes.
In 1998, Wills (pp. 205, 208) pointed out:
Separating the important from the unimportant changes in our DNA will soon be possible. … [O]ver the next few decades, we will be able to compare all our DNA with that of chimpanzees and gorillas and thereby track down the many differences. … A small number of … highly significant regulatory bits of our genomes must have been changing at top speed over the last few million years. We suspect, without direct proof as yet, that fewer such changes have happened in the chimpanzee lineage. … The trick will be to track down the changes that demonstrate this.
As this review will show, at least some of these predictions are turning out to be correct.
Because of new DNA evidence, the question of whether our evolution is continuing has now been answered in the affirmative (López Herráez et al., 2009; Nielsen et al., 2007; Pickrell et al., 2009; Xue et al., 2009). But an equally important question remains to be addressed fully. What is the nature of the variation that has accumulated in our gene pool and that permits this rapid evolution?
This review will examine the growing evidence for the shaping of our gene pool through natural selection and explore some of the less obvious consequences of these discoveries. In particular, it will ask whether there is evidence that negative frequency-dependent selection (Ehrman, 1970) is helping to shape our genomes. Negative frequency-dependent selection takes place when rare alleles, phenotypes, or species have a selective advantage that disappears or even reverses when these alleles, phenotypes, or species become more common. A classic example in Drosophila is the rare-male effect (Ehrman, 1970) in which mutant males have a mating advantage when they are rare and a disadvantage when they are common. In humans, frequency-dependent selection is seen in the selection of rare alleles at the major histocompatibility complex (HLA complex in humans) by continually shifting sets of pathogens (Borghans et al., 2004).
Nature of DNA Sequence Data
The first extensive survey of genetic variation within a species at the DNA level was carried out by Kreitman (1983), using the alcohol dehydrogenase gene of Drosophila melanogaster. The amount of sequence generated was by the standards of the time monumental—a stretch of 2688 bases of DNA that spanned the entire gene, sequenced from 11 different flies. This stretch of DNA included all the exons and introns and substantial portions of the flanking 5′ and 3′ regions of the gene.
A quarter of a century later, of course, much has changed. The total of 10,358 bases that Kreitman sequenced over months of demanding work would represent only 0.00004% of the daily output from current top-of-the-line sequencing machines (see for example http://www.illumina.com/systems/hiseq_2000.ilmn#workflow_spec).
In his sample of genes, Kreitman found only one nonsynonymous substitution, that is, a substitution that codes for an amino acid change in the protein. This substitution was an A–C transversion, in which the purine base A had been substituted for the pyrimidine base C, or vice versa. This DNA polymorphism resulted in a threonine–lysine protein polymorphism. This polymorphism in turn defined fast and slow alleles that had already been identified by the rates of migration of the ADH proteins in an electrophoretic field.
Kreitman also discovered a pattern of strong linkage disequilibrium at the ADH locus. Two distinct clusters of differences that had no effect on the protein were clearly linked to the two differences that did have an effect (Fig. 1).
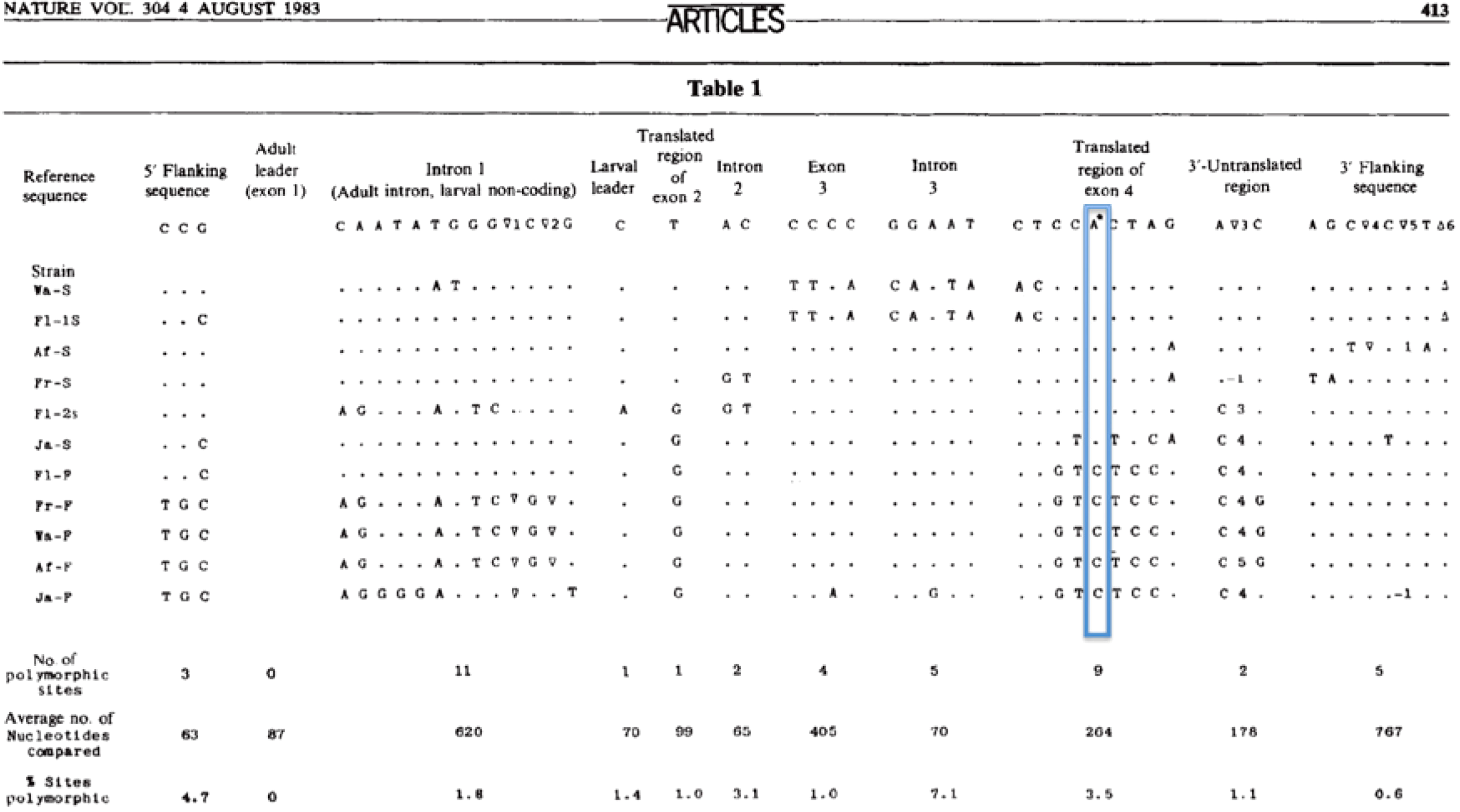
Kreitman's Table 1. The table in Fig. 1 shows only the sites at which differences were found among the 11 alleles. The table has been slightly modified with the addition of a box to draw attention to the single nonsynonymous difference between the six slow and five fast alleles. Note that the fast and slow alleles carry synonymous polymorphisms (one in exon 2 and six in exon 4) and silent polymorphisms (such as the three in the 5′ flanking sequence and the ten in intron 1) that are in strong linkage disequilibrium with the fast and slow alleles. Note also that there seems to be stronger disequilibrium of these polymorphic sites among the fast alleles than among the slow alleles. Such a pattern can be interpreted as evidence for selective sweeps involving the nonsynonymous site, with the fast allele possibly arising and being swept to high frequency more recently.
One way of interpreting the pattern that Kreitman found is that it provides evidence for selective sweeps (Aguade et al., 1988; Kaplan et al., 1989) that have driven both the fast and slow alleles to high frequencies in the Drosophila population. Such sweeps may be the result of unconditional advantage to the favorable allele or conditional advantage of the heterozygote balanced by disadvantage of the homozygotes, such as has been demonstrated in the sickle-cell allele (Aidoo et al., 2002) and suspected for mutations causing cystic fibrosis (Alfonso-Sanchez et al., 2010).
When the original mutant fast and slow alleles began to spread, they carried along the linked genetic differences that happened to be present on the chromosomes on which they first arose. The sweeps are likely to have been relatively recent; otherwise, genetic recombination would have separated each allele from its accompanying set of synonymous and silent changes. Indeed, you can see some examples of recombination in Kreitman's Table 1. Sequences F1-2s and Ja-S have picked up some of the bases that accompanied the fast allele.
Now that it is possible to compare entire genomes, patterns indicative of possible selective sweeps are found scattered throughout the human gene pool (Sabeti, 2006). Some of the most informative of these patterns show stronger amounts of disequilibrium on some chromosomes than on others. Such discordant patterns of linkage disequilibrium are unlikely to result from random drift and/or localized differences in recombination rate, which would affect all the chromosomes in the population equally.
Most mutational changes are either selectively neutral or disadvantageous. The disadvantageous alleles, removed by purifying selection (Kimura and Ohta, 1974), will drag neutral or even some advantageous linked alleles with them as they disappear from the population.
All measures of human evolutionary change show a large role for such purifying selection. Estimates of the fraction of DNA sequence sites affected by purifying selection have grown during the last decade from 37% (Eyre-Walker and Keightley, 1999) to 75% or more (Kryukov et al., 2007).
If many disadvantageous alleles arise in a region of a chromosome, it will be emptied of polymorphism, slowing its rate of evolutionary change. But there are alternative explanations for slowly evolving regions. It is possible that they simply have a low mutation rate. Or they may have a low rate of recombination so that accumulating harmful alleles that appear near advantageous linked alleles can remain linked to them for long periods and drag even these positive alleles down to the point where they disappear from the gene pool. Complicating matters further, random drift and gene flow between populations can sometimes imitate the signatures of both directional and purifying selection (Nielsen, 2001).
Regions of the Genome That Have the Potential for Selection
Until relatively recently, most hunts for the effects of selection have concentrated on translated parts of the genome, in particular on the relative rates of evolution of nonsynonymous changes in the code that bring about changes in amino acid sequence and synonymous changes that leave the amino acid sequence unaltered. Hudson and colleagues (1987) and McDonald and Kreitman (1991) pointed out that if the amount of between-species nonsynonymous change is less than would be expected, given the level of within-species polymorphism, then purifying selection has acted to purge variants from the population. At the other extreme, if between-species nonsynonymous changes are commoner than would be expected from the amount of polymorphism that has accumulated within the species, then there has been directional selection in the course of species divergence. These expectations led to a family of tests for selection, known as the Hudson–Kreitman–Aguade
Nonsynonymous changes, such as the ADH fast and slow variants, are more likely than synonymous changes to be subject to both purifying and directional selection (Fay et al., 2002). When these two conflicting types of selection act on linked sites within small segments of the genome, they may cancel out each other. Thus, even if genes are strongly selected, signs of their selection might be missed when McDonald–Kreitman and similar measures are used.
Since the invention of these early tests, much has changed in our understanding of the genome. Regulatory regions upstream and downstream of genes are common, as are enhancer regions that can be tens of thousands of bases away—or even on different chromosomes—from the gene that they affect. Interactions between genes and their regulatory regions give rise to what has been called the transcriptional interactome (Schoenfelder et al., 2010). Alternative transcription and alternative splicing, which allow the same gene to take up different functions in different tissues, have now been shown to affect the majority of eukaryotic genes (Hallegger et al., 2010). And the definition of regulation has broadened dramatically to accommodate the discovery of a growing zoo of functional RNAs that are highly conserved during evolution (Liu and Paroo, 2010). Perhaps most remarkably, the majority of the human genome turns out to be transcribed, providing enormous opportunity for levels of regulation that are still being discovered (Lindberg and Lundeberg, 2010).
Little is known about the effect of natural selection on these regions of the genome outside genes, although, as noted above, it is clear that many untranslated regions have been highly conserved during primate evolution. Given these levels of regulatory complexity, it is not surprising that some regions of the genome are likely to be more sensitive to evolutionary change than others (Fay et al., 2002). It has become apparent (at least in Drosophila) that synonymous changes within genes are less subject to purifying selection than so-called “silent” changes that lie in regions between the genes (Andolfatto, 2005). As advances in our understanding of gene regulation would predict, these between-gene (Ohno, 1972) regions are actually so important to the organism's development that changes in them are selected against. There is also some evidence from Drosophila that there has actually been positive selection on some of these same regions that lie between the genes (Andolfatto, 2005; Sella et al., 2009). These islands of positive selection seem likely to mark the location of regions that regulate the expression of protein-coding genes. If species are adapting to new environmental circumstances, many such regions might be expected to undergo rapid evolution.
The strength of genetic recombination varies in different parts of the genome and can change as species diverge (Dumont and Payseur, 2008). If levels of recombination are high, they would be expected to increase the rate of directional selection, because high rates of crossing-over allow beneficial alleles to break free of linked deleterious alleles and spread more easily in populations. Low levels of recombination, on the other hand, might slow evolutionary change because of the fact that harmful mutations always tend to outnumber beneficial ones (Nielsen et al., 2007). If harmful alleles remain linked to new beneficial alleles for many generations, they can drag the beneficial alleles down with them. Signals of selection could be lost under such circumstances (Nielsen et al., 2007).
The role of balanced polymorphisms has received little attention in models that are based on the McDonald–Kreitman and HKA approaches. Balancing selection has maintained some polymorphisms for periods much longer than the periods of existence of the species that currently carry them. These include the so-called transspecific polymorphisms such as allelic variants at the human lymphocyte antigen (HLA) system that is essential for the adaptive immune system (Klein et al., 2007), and at the antiviral gene TRIM5 that interferes with the replication of some retroviruses (Johnson and Sawyer, 2009). If the selective pressures remain the same as species diverge, these regions may undergo little change and be missed by algorithms searching for signs of selection. Thus there may be regions of the genome that are actually being subjected to strong and complex selection pressures, but the commonly used statistical methodologies for making between-species comparisons may not be able to pick them up because they assume that only purifying or directional selection are operating.
Methods for Measuring Evolutionary Change
Various methods that can disentangle the processes that bring about evolutionary change have been proposed. Their ability to avoid false positive and false negative signals of selection depends on the assumptions that underlie them and on the type, extent, and quality of the available sequence data.
The hunt for genes that cause disease has greatly accelerated our ability to find genetic variants within the human gene pool. This intensive search has produced astounding new technologies for sequencing DNA, including the ability to sequence modified epigenomes (Fazzari and Greally, 2010). This dizzying new technology has provided population geneticists with a body of information about our species that could never have been obtained if it was not of inestimable value to the entire biomedical community. As a by-product of the massive searches for such biomedical advances as new therapies for cancers, the availability of multiple genomic sequences has had the fortuitous effect of enabling us to find traces of natural selection in our genomes.
Even more luckily from the standpoint of evolutionary biology, the incentives for gathering further masses of information about our gene pool are strong. The first surveys of our gene pool for genetic polymorphisms that could aid in the hunt for association of diseases with genetic markers concentrated on linked groups of markers (haplotypes) that are found at high frequencies in various human groups. Such haplotypes are relatively easy to find, because they are present in a substantial number of individuals and are therefore likely to turn up in small samples of genomes. But the first attempts to associate such common haplotypes with disease have generally been disappointing. Associations tend to be weak even with diseases that are known from twin and family studies to have high heritability.
Several explanations have been proposed to account for this “missing heritability” (Maher, 2008). The most likely possibility is that many high-heritability diseases can be caused by homozygosity for rare genetic variants at one or more of many different loci. This likelihood is increased by the finding that all humans are carriers of loss-of-function alleles at 2 to 300 loci (Thousand Genome Consortium, 2010).
If this model for harmful genes in our gene pool is correct, the hunt for disease genes requires the accumulation of enormous numbers of highly accurate haplotypes, including those that carry rare variants, from many different human genomes. The numbers involved are likely to be huge. The common variants that were collected in the original HapMap project are estimated to top out at about 10 million and to be involved in several hundred thousand common haplotypes. The newer Thousand Genomes Project (Thousand Genome Consortium, 2010) has already found 15 million mostly new single-nucleotide polymorphisms, detected by low-coverage sequencing of 179 genomes and high-coverage sequencing of two small families (Thousand Genome Consortium, 2010). These new polymorphisms include essentially all those that are present in 10% or more of the sampled chromosomes, but the number of polymorphisms at frequencies between a fraction of a percent and 1% that are present in the entire human gene pool is likely to be in the billions.
Various statistical tests can be used to compare sets of genomes that have such a high density of information. One of the simplest is the Tajima (1989) D, which compares the polymorphism at a site in two or more different subpopulations with neutral expectation. If the populations have diverged entirely as a result of selectively neutral changes, the ratio of fixed differences to polymorphisms can be predicted. If there are unusual numbers of polymorphisms at low frequency, this may indicate purifying selection or population expansion or both. Unusual numbers at high frequency may indicate balancing selection or population shrinkage or both. Because deviations from neutrality detected by this statistic have more than one possible cause, they are difficult to interpret.
More specific tests that can be applied to moving-window surveys of entire genomes hold more promise. The locus-specific branch-length test (Messier and Stewart, 1997) and its many variants (Yang and Dos Reis, 2010) look for differences in the branch lengths of trees built from the polymorphisms found in the subsets of data. If the branch lengths of a given region of the genomes being compared are longer or shorter than average, this suggests that there have been unusually large or small numbers of substitutions or allele frequency shifts in that region, suggesting purifying or directional (and/or balancing) selection, respectively. The long- range haplotype test (Sabeti, 2006), and its extension, the whole-genome long-range haplotype test (Zhang, 2006), examine unusual cases of linkage disequilibrium that reveal the selective history of alleles at a locus. In this test, the pattern looked for is an unusually high level of linkage disequilibrium involving only some of the haplotypes that are being compared. Those haplotypes must therefore have been increased in frequency recently, probably by directional selection.
One of the largest such surveys to date (Hawks et al., 2007) used HapMap data consisting of 4 million single-nucleotide polymorphisms to find 2300 such selected regions in a Chinese dataset, 2800 each in European and Japanese datasets, and almost 3400 in an African (Yoruba) dataset. Strikingly, only about 500 of these large numbers of incomplete selective sweeps were shared between datasets. Even more strikingly, there were almost no parts of the genome in any of the datasets that had clearly swept to complete fixation.
Hawks and colleagues (2007) argue that the paucity of completely fixed regions and the relatively large size of the chromosome regions in disequilibrium means that this wave of selection has been recent and strong, starting about 80,000 yr ago in the African population and about 40,000 yr ago in non-African populations. They also argue that these waves of selection are highly unusual in our species. If such strong selection had continued for longer periods, the amount of heterozygosity in the human genome would be much lower because of many completed selective sweeps.
Comparisons of genome sequences that have changed between humans and other primates have picked up a large number of highly suggestive regions in which there is some evidence for directional selection. These results, however, are intensely dependent on the methodology and the databases that were used. Table 2 (Nielsen et al., 2007) summarizes the low concordances among several such studies, suggesting that the majority of the results so far, while interesting, are in desperate need of confirmation using more extensive databases.
Recent Rapid Directional Evolution
The race to find genetic correlates of disease ensures that the amount of genetic information available to evolutionary biologists will continue to grow rapidly. As an incidental result of these new technologies, for the first time it is becoming possible to examine the evolution of different human groups that have been subject to extreme environments.
Several human and hominid groups have moved to high latitudes in Europe and Asia. It is now clear, from examination of genes that are known to be involved in pigment production, that Neanderthals and modern Europeans independently acquired different mutations in the melanocortin-1 gene that affects skin and hair color (Lalueza-Fox et al., 2007). Asians have acquired light skin color, and presumably a greater ability to use sunlight to manufacture vitamin D, through different mutations in genetic pathways that they share with Europeans and in different pathways (Norton et al., 2007). Lactose tolerance that lasts throughout life and that allows lifelong utilization of dairy products has evolved swiftly in Europeans (Bersaglieri et al., 2004). The lactase enzyme has evolved toward lifelong expression independently in Europeans and in East Africans (Tishkoff et al., 2007). And we may have accelerated the evolution of genes in the animals on which we depend. Milk protein genes in cattle have also evolved rapidly during the last 7000 yr in Europe (Beja-Pereira et al., 2003). These surveys have examined genes that were already strongly suspected to play a role in adaptation. In all cases, it should be noted, the mutant alleles have not swept to fixation in the selected populations.
Several groups have looked for and found recent selection at loci in human populations that are likely to increase adaptation to high altitude (Beall et al., 2010; Bigham et al., 2010; Bigham et al., 2009; Simonson et al., 2010; Yi et al., 2010). There are strong indications of selection acting on genes that decrease hemoglobin production in Tibetans and on genes of the hypoxia inducible transcription factor pathway in both Tibetan and Andean populations. The apparently most strongly selected allele, a substitution in the gene EPAS1, has moved to near-fixation in Tibetan populations (Yi et al., 2010). The authors' calculations (Aldenderfer et al., 2011) (Shi et al., 2008). that Tibetan and Han populations diverged only 2750 years ago are likely to be in error because of evidence for long-continued human occupation of the Tibetan plateau. Even if these estimates prove to be correct, the spread is not much faster than the spread of lactose tolerance in European populations and would have been aided by the initial small size of Tibetan populations and strong selection that has acted on high altitude populations (Mazess, 1965).
Selection for Polymorphism
The current major groupings that make up the human HapMap set of data are African (chiefly Yoruba at the moment), European, Chinese, and Japanese. It is striking that very few complete allelic substitutions have taken place during the time that these groups have diverged (Coop et al., 2009). In addition to the obvious likelihood that there has been continuing admixture between the populations, there are also two nonexclusive selective explanations for this pattern. First, there may simply not have been enough time for substitutions to go to fixation. This would particularly be the case if the alleles that are undergoing substitution are dominant so that their rate of fixation will be slowed when most of the remaining alternative alleles are heterozygous and no longer being selected against. Second, many selected alleles may not be unconditionally advantageous and may lose their advantage as they increase in frequency. This has been the case, for example, with HLA (human MHC) alleles, which only have a strong advantage when they are rare (Borghans et al., 2004). Evidence is growing that as HLA alleles increase in frequency they lose their advantage as the mix of pathogens they are defending against changes. They will thus reach intermediate frequencies without going to fixation.
As noted earlier, Hawks and colleagues (2007) found evidence for about 500 regions in the human genome in which polymorphisms have changed little in frequency among African, European, and Asian subpopulations. It appears that these regions, which make up about 15% of the selected regions in their study, have maintained their polymorphisms by balancing selection. Many other balanced polymorphic alleles may have been missed, however, in cases in which the establishment of new polymorphisms involved shifts in the frequencies of alleles to new equilibria.
When extensive data from groups of Africans other than the Yoruba become available, it will be extremely instructive to see whether incomplete substitutions are also characteristic of the differences between these African lineages. Some African groups, notably the San, pygmies, and other hunter–gatherers such as the Hadza, have diverged for longer periods than the groups that left Africa for Europe and Asia (Behar et al., 2008). If there are large numbers of incompletely fixed substitutions even among human groups that have been separated for 100,000 yr or more (Ingman et al., 2000), this finding will provide a strong indication that selection for balanced polymorphisms has played and is continuing to play a large role in the evolution of different human groups.
An alternative possibility is that gene flow has been able to take place even between apparently long separated populations. The possibility of gene flow should be distinguishable from that of balancing selection if enough information is available about complete genomes in the populations between which the putative gene flow may have taken place.
At the moment, most of the evolutionary changes that are known to have taken place in humans involve changes in physiology. As our knowledge of gene function grows, we will find more changes that have had an effect on brain function. This author has made some predictions concerning the nature of the accumulated variations and the possible role of negative frequency dependence in the maintenance of genetic diversity.
Might the phenomenon of frequency-dependence help maintain and even increase human behavioral diversity? One fascinating and little-noticed aspect of frequency-dependent selection is that it reinforces itself. … Thus diversity itself will be selected for. … The opportunities for human behavioral diversity are greater than they have ever been. As a result, more and more of our underlying genetic diversity is revealed. (Wills, 1998)
Our species is marked by far more behavioral polymorphism than any other species (Gardner, 1993). The opportunities for expressing and benefiting from this behavioral diversity are growing as our cultures become more complex. Searches for signs of recent human evolution should keep an open mind about the possibility that, because of this growing behavioral diversity, there may be a major evolutionary trend toward the establishment of new polymorphisms that are being maintained by negative frequency-dependent selection.
Regardless of whether alleles of genes regulate physiology or behavior, they are subject to the same set of selective pressures. At the moment, it is impossible to say what fraction of such substitutions currently taking place in our genome are moving toward complete fixation, perhaps hindered by gene flow, and what fraction are moving toward an internal equilibrium. But it is worth noting that the genetic load or burden of mortality that is associated with the maintenance of large numbers of polymorphisms by negative frequency-dependent selection can be low as long as the polymorphic alleles are near their internal equilibria (Wills, in preparation). This is because, unlike selection for heterozygote advantage, the strength of selection decreases as frequency-dependent polymorphisms approach their equilbria. Thus, there is no theoretical impediment to the accumulation of large numbers of this type of balanced polymorphism in our species.