
Abstract
Abstract
Abeysekera, W.Y.M., W.D.D. de Silva, S.S. Pinnaduwa, and A.S.K. Banagala. Acute Massive Splenic Infarction with Splenic Vein Thrombosis Following Altitude Exposure of a Sri Lankan Male with Undetected Sickle Cell Trait. High Alt Med Biol 13:288–290, 2012. —Even though sickle cell disease is not common in Sri Lanka, we report an acute splenic infarction at high altitude of a Sri Lankan male with previously undetected sickle cell trait (SCT). This is the first time such a case is reported from the South Asian region. Early recognition of this hematological condition would simplify the management of acute splenic infarction in these patients, avoiding irreversible surgery.
Introduction
Case Report
A 31-year-old tailor from Ambalanthota in the Hamanthota district presented to our surgical casualty with acute left upper abdominal pain with vomiting at the peak of ‘Sri pada’ (Adam's peak), which is a 7360 ft high sacred mountain in Sri Lanka. He was admitted to our casualty 2 days after the onset of symptoms due to lack of response to medication. This was his fourth journey to the same mountain during the past 10 years. Except for mild abdominal pain and few episodes of vomiting that settled spontaneously in his third climb, he was completely healthy in previous trips.
On examination, he had a tender splenomegaly, with ultrasound evidence of a massive splenic infarction with splenic vein thrombosis. Abdominal CT further confirmed an enlarged spleen with massive splenic infarction (Fig. 1). Only a small central portion, 3.5 cm x 1.8 cm, of spleen showed enhancement with contrast in arterial phase. There was also evidence of thrombosis of the distal part of the splenic vein, with normal inferior mesenteric vein, superior mesenteric vein, portal vein, and splenic artery.
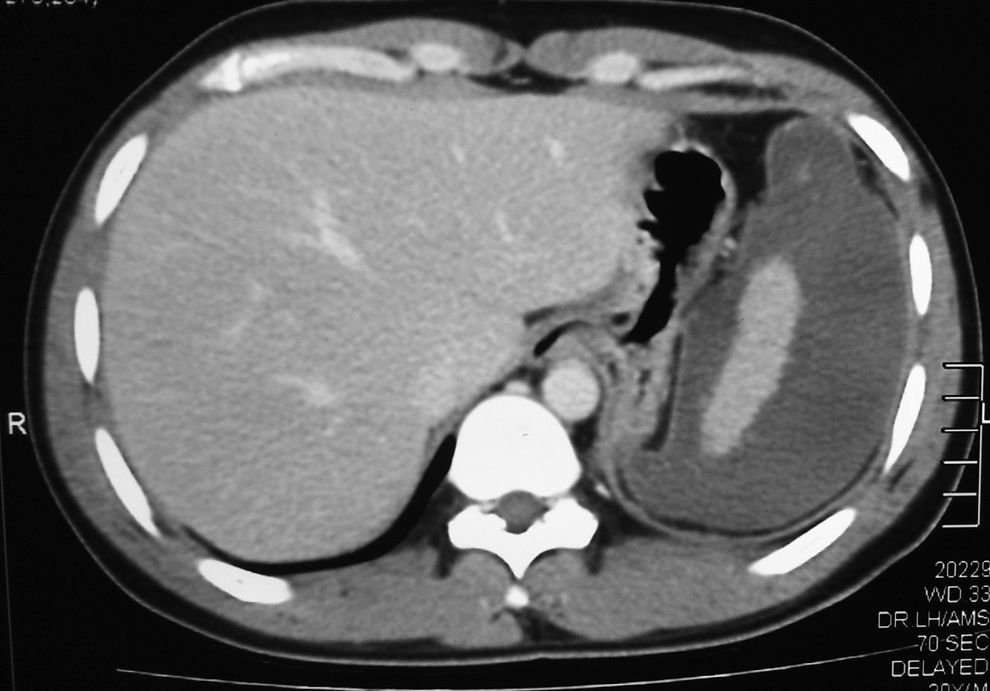
CT demonstrating the massive infarction of the spleen.
FBC revealed a WBC 11300/μL, Hb 12.1 g/dL, MCV 86.7 fL, thrombocytosis (657,000/μL), but the blood pictures were suggestive of an infective or inflammatory process due to neutrophil leukocytosis with a left shift. Except for the marginally elevated total bilirubin level (22 μmol/L), the liver profile was normal. As suggested by the hematology team, high performance liquid chromatography (HPLC) was performed which demonstrated HbA-49.3%, HbA2-3.1%, HbF-0.9%, and HbS-42.6%. HPLC findings with the positive sickling test confirmed the sickle cell trait (SCT) of our patient.
He completely recovered on conservative management with adequate hydration, analgesics, and oxygen therapy. After immunization for capsulated organisms, the hematology team planned for long-term folate therapy and further follow-up for his hematological condition, including family screening for the sickle cell gene.
Discussion
Sickle cell traits (SCT) are highly prevalent among Black populations in Africa and African-Americans in the United States. As the most common hemoglobinopathy in the world, it is not uncommon in other regions such as Latin America, the Middle East, and Asia. In India, sickle cell disease (SCD) was first detected among tribal groups in south India (Lehmann and Cutbush, 1952), and now it is identified as a wide-spread condition where the prevalence may reach 9%–22% among different castes and communities in India (Shukla and Solanki, 1985).
In 1962, De Silva et al. reported their investigations of a Sinhalese family with HbS from Eastern province in Sri Lanka where the gene was detected in 3 generations of the family. In 1989, Nagaratnam determined that the Sri Lanka Hambantota district had the highest incidence of the HbS gene. The different ethnic groups in Sri Lanka vary in the incidence of abnormal HbS, and migration and natural selection must be the most reasonable explanation for that. Our patient is from a village in the Hambantota district in Sri Lanka. The first homozygous sickle cell disease in Sri Lanka was reported by Lucas and Jayawardena (1991), but the real picture of the prevalence of SCD in Sri Lanka is yet to be studied.
At high altitude, the hypoxic environment may induce the proliferation of hemoglobin S (HbS) and deformation of red cells, which will lead to acute sequestration of sickled red cells in the spleen and vaso-occlusion. Patients develop acute left hypochondriac pain with anorexia and vomiting with guarding and severe tenderness over the splenic area. This is known as acute splenic sequestration syndrome, which may lead to an acute splenic infarction (Goldberg et al., 1985; Sheikha, 2005; Chamberland, 2006; Morishima et al, 2008). Our patient most likely experienced an acute splenic sequestration syndrome without a splenic infarction in his third visit to the sri pada, but in his last visit the splenic syndrome resulted in a splenic infarction.
Sheikha (2005) identified 47 published cases of splenic infarctions at high altitude from 1985 up to 2005, and all but one of them were male patients. Lane and Githens (1985) explained that SCT may develop acute splenic sequestration or splenic infarction when exposed to moderate to high altitudes (5000 ft—10,000 ft). Thus, they suggested that after exposure to high altitudes greater than 5000 ft, if someone develops significant left upper abdominal pain, splenic syndrome or splenic infarction must be considered, regardless of the patient's race, because he or she may be a person with undetected SCT. This was true in our patient since he became symptomatic at the peak of the mountain (nearly 7000 ft) and he was not from a community where the SCD is prevalent.
Some authors hypothesized that the level of HbS percentage is a predisposing factor for splenic syndrome or infarction, and a level more than 40% was considered to be a higher level that can be seen only in one-third of SCT individuals (Goldberg et al., 1985; Lane and Githens (1985). But others suggest the presence of a compound heterozygous state such as Hb SC or sickle-thalassemia trait would be a more important risk factor than the level of HbS (Sheikha, 2005). Our patient had an HbS level of 42.6% but there was no evidence of a compound heterozygous state. However, it is well observed that patients with sickle cell anemia (HbSS) are protected from acute splenic infarction at high altitude due to autosplenectomy, which may happen early in life due to repeated vaso-occlusive crisis (Goldberg, 1985).
Splenic vein thrombosis is another recognized cause of splenic infarction (Gupta and Kakar, 2004). But the splenic vein thrombosis in our patient is probably secondary to splenic infarction since he had clear evidence of a well-recognized cause.
Treatment for splenic infarction in these patients should be mainly conservative. In many instances, unnecessary splenectomy was performed due to unawareness of the sickle cell state of the patient. Literature strongly supports supportive therapy with adequate hydration, analgesics, and oxygen therapy as the main steps of treatment without any surgical intervention (Lane and Githens, 1985; Sheikha, 2005).
As evident in the literature, splenic infarctions induced by altitude hypoxia in SCT are not always limited to regions where the SCD is common (Sheikha, 2005). Therefore, the possibility of undetected SCT condition should be considered as a differential diagnosis in splenic infarctions following altitude exposure, even in a region with low or undetermined prevalence for SCD.
Footnotes
Author Disclosure Statement
No competing financial interests exist.