
Abstract
Abstract
Fan, Jui-Lin, and Bengt Kayser. Repeated pre-syncope from increased inspired CO2 in a background of severe hypoxia. High Alt Med Biol. 15:70—77, 2014.—We describe a case of experimentally induced pre-syncope in a healthy young man when exposed to increased inspired CO2 in a background of hypoxia. Acute severe hypoxia (FIO2=0.10) was tolerated, but adding CO2 to the inspirate caused pre-syncope symptoms accompanied by hypotension and large reductions in both mean and diastolic middle cerebral artery velocity, while systolic flow velocity was maintained. The mismatch of cerebral perfusion pressure and vascular tone caused unique retrograde cerebral blood flow at the end of systole and a reduction in cerebral tissue oxygenation. We speculate that this occurrence of pre-syncope was due to hypoxia-induced inhibition of brain regions responsible for compensatory sympathetic activity to relative hypercapnia.
Introduction
S
Syncope is well documented during acute exposure to hypoxia (Nicholas et al., 1992; Sagawa et al., 1997; Westendorp et al., 1997; Kapoor, 2002; Roche et al., 2002; Van Lieshout et al., 2003; Halliwill and Minson, 2004), while high altitude acclimatization appears to be rather protective against syncope at high altitude (Thomas et al., 2010). In this case report, we present novel data from two trials of acute exposure to severe hypoxia in combination with relative hypercapnia. We observed pre-syncope symptoms accompanied by hypotension, cerebral hypo-perfusion, and cerebral tissue desaturation in a 21-year old male, when inspired CO2 tension was increased in a background of hypoxia equivalent to an altitude of 5000 m.
Methods
Subject details
The subject was one of a group of volunteers participating in a study designed to examine the effect of inspired CO2 on exercise capacity in hypoxia. He was a 21-year-old healthy nonsmoking physically active male (BMI=23.2, V
Consent
Written informed consent was obtained from the subject for publication of this case report.
Measurements
The following variables were recorded continuously: bilateral systolic, diastolic and mean middle cerebral artery velocity (MCAv, transcranial Doppler ultrasound, ST3, Spencer Technology, Seattle, USA; monitoring probe (2 MHz) power: 50%, depth: 57 mm); heart rate (HR), cardiac output (Q’), stroke volume (SV), total peripheral resistance (TPR), beat-to-beat systolic, diastolic and mean arterial blood pressure (ABP, finger plethysmography, Finometer-MIDI, Finapress Medical Systems, Amsterdam, Netherlands); peripheral O2 saturation (Sp
Experimental protocol
The original protocol foresaw, with the subject seated on a cycle-ergometer, 4 min of normoxia baseline measurements, 2–3 min of hypoxia, followed by 4 min of increased inspired CO2, to bring P
Throughout the experimental sessions, the subject wore a nose-clip and breathed through a mouthpiece attached to a low resistance one-way non-rebreathing valve (Hans-Rudolph 2700, Kansas City, USA). The hypoxia and added CO2 to the inspirate were achieved using a modified gas mixing system (Altitrainer, SMTec, Nyon, Switzerland), described in detail by Fan et al., (2013). For this experiment F
Results
The subject attended the laboratory on four occasions. On the first two visits, he successfully completed incremental maximal cycling tests (30 w/min ramp) in conditions of normoxia and normoxia+CO2 without any adverse events. On the third occasion, the subject was seated on the cycle ergometer in preparation of the next test, breathing room-air for 4 min. He was then switched to hypoxia for 3 min, followed by increased inspired CO2 in hypoxia. After 134 sec, pre-syncope symptoms prompted the subject to come off the mouthpiece and request the session to be aborted. The subject reported symptoms of dizziness, blurry vision, and nausea. We invited the subject to come back to the laboratory on another occasion, this time adding the measurement of oxy- (O2Hb) and deoxygenated hemoglobin (HHb) over the left frontal cortex with near-infrared spectroscopy (index of brain tissue oxygenation; NIRS, Oxymon Mk-II, Artinis, Zetten, Netherlands), expressed as absolute changes from baseline room-air breathing. The experiments were performed at the same time of the day. On the second occasion, the subject was left on hypoxia for more than 4 min before switching to increased inspired CO2 to exclude that he was not simply developing a vasovagal reaction to hypoxia. The subject indicated that he was fine, after which he was switched to added CO2. Following 55 sec of breathing CO2-enriched hypoxic gas, the subject indicated that pre-syncope symptoms were developing again and came off the mouthpiece, requesting the experiment to be aborted. We then excluded the subject from further participation.
Table 1 shows the values for the measured and derived variables during the different epochs of the two episodes of hypoxic+CO2, as well as the data from the normoxic+CO2 visit for reference (means over 20 sec at the end of each epoch). Hypoxia alone lowered P

Cerebrovacsular and cardiovascular variables during visit one. Middle cerebral artery velocity (MCAv), cerebral vascular resistance (CVR), arterial blood pressure (ABP), total peripheral resistance (TPR), cardiac output (Q’), stroke volume (SV), heart rate (HR), and peripheral O2 saturation (Sp
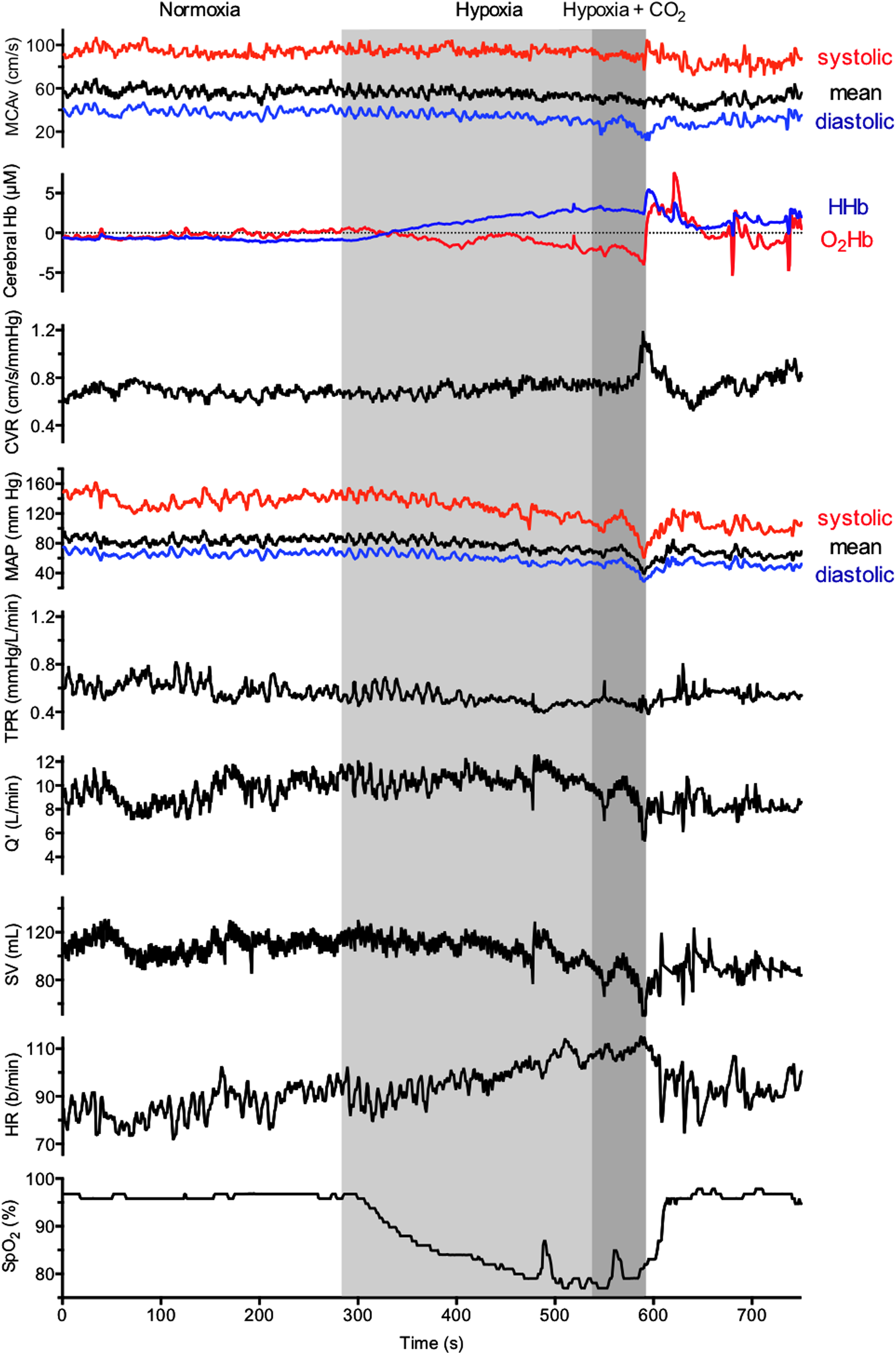
Cerebrovascular and cardiovascular variables during visit two. Middle cerebral artery velocity (MCAv), cerebral hemoglobin concentration (Hb), cerebral vascular resistance (CVR), arterial blood pressure (ABP), total peripheral resistance (TPR), cardiac output (Q’), stroke volume (SV), heart rate (HR), and peripheral O2 saturation (SpO2).

Middle cerebral artery velocity traces during hypoxia
n.m.: not measured; o.m.: off mouth piece.
Discussion
We report the sudden development of pre-syncope symptoms in a healthy 21-year old male during acute exposure to inspired CO2 in a background of severe hypoxia. The pre-syncope symptoms were accompanied by bradycardia, hypotension, cerebral hypo-perfusion, and cerebral tissue deoxygenation. The observed changes in vital signs, HRV index LF/HF, and development of symptoms seems coherent with a vasovagal reaction.
The mechanisms involved in acute hypoxia syncope are complex. The increased susceptibility to syncope in hypoxia has been attributed to a reduced baroreflex sensitivity (Nicholas, 1992; Sagawa et al., 1997; Roche et al., 2002; Klemenc and Golja, 2011), which is further compounded by greater cardiac output decline and withdrawal of sympathetic vasoconstrictive tone during orthostatic stress in hypoxia, resulting in greater hypotension (Wang et al., 1996; Van Lieshout et al., 2003). Westendorp et al., (1997) proposed that an enhanced vasodilatory response to epinephrine during hypoxia (Blauw et al., 1995) would lead to greater sympathetic withdrawal as well as elevated parasympathetic activity. They further proposed that an increase in atrial natriuretic peptide during hypoxemia might attenuate the carotid baroreflex-mediated cardiac acceleration and inhibit sympathetic nervous activity, thus further increasing the risk of syncope in hypoxia.
To the best of our knowledge, this is the first report documenting reduced CBF during a pre-syncope response triggered by CO2-enriched inspired gas in a background of hypoxia. The observation during the second episode of a drop in frontal cortex oxygenation suggests that the pre-syncope syndrome was accompanied by reduced cerebral oxygen availability (Fig. 2). We found dramatic reductions in mean and diastolic MCAv during and immediately following hypoxia+CO2 exposure (Fig. 1). Meanwhile, the reduction in cerebral O2Hb followed closely to the changes in diastolic MCAv (Fig. 2), suggesting that diastolic perfusion pressure is the main determinant of cerebral O2 delivery. Vasovagal syncope typically involves both central and peripheral mechanisms. However, since severe and uncompensated hypotension, below the threshold of cerebral autoregulation, leads to cerebral hypo-perfusion ( Skinhoj and Strandgaard, 1973; Chillon and Baumbach, 1997), while hypotension per se blunts the CBF responsiveness to CO2 during both normoxic and hypoxic conditions ( Harper and Glass, 1965; Ainslie et al., 2012), we attribute the observed reduction in CBF and subsequent cerebral tissue deoxygenation to the direct effect of severe hypotension per se.
According to Van Lieshout et al., (2003), when a pre-syncope progresses, it is the fall in mean ABP below the lower limit of cerebral autoregulation that reduces cerebral perfusion and oxygenation, leading to unconsciousness. In the present case, we observed dramatic reductions in mean ABP with increased inspired CO2 in background hypoxia, which persisted for some time following the exposure (Fig. 1). In addition to this drop in ABP, we observed a distinct triphasic flow pattern in the CBF velocity waveform, characterized by large flow during systole, followed by a distinct retrograde flow at the end of systole, and very little flow during diastole (Fig. 3). Since CO2 is a potent vasodilator of the cerebral vasculature, we reasoned that the reduction in mean ABP associated with increased inspired CO2 would lead to a greater mismatch of cerebral perfusion pressure and vascular tone (i.e., impairment of cerebral autoregulation). This mismatch of cerebral perfusion pressure and vascular tone could potentially account for the retrograde flow observed in our subject.
What are potential mechanisms for such an effect of increased inspired CO2 in a background of hypoxia? In normoxic conditions, hypercapnia has been shown to elicit a vasodilatory effect on the peripheral vasculature in humans (Lennox and Gibbs, 1932; Kontos, 1971; Kontos et al., 1972; Gastaldo et al., 1974; Ainslie et al., 2005). Hypercapnia (10% CO2) causes an immediate, but transient hypotension, which is corrected within 30–40 sec from increased sympathetic activity, in anesthetized, sino-aortic denervated and vagotomized rats (Takakura et al., 2011). Since inhibition of the retro-trapezoid nucleus (RTN) lowers sympathetic nerve activity and attenuates mean ABP recovery at the end of the hypercapnic exposure (Takakura et al., 2011), those authors concluded that the compensatory increase in sympathetic nerve activity is partly dependent on RTN activation during hypercapnic exposure. Similarly, pontine noradrenergic neurons, astrocytes, neurons of the nucleus solitary tract, and wake-ON orexinergic neurons, have all been shown to be hypercapnia/pH-sensitive and linked to sympathetic tone, contributing to peripheral vascular tone during hypercapnic exposure (Dean et al., 1989; Dun et al., 2000; Johnson et al., 2008; Allen and Barres, 2009). In humans, profound sympatho-inhibition has been reported during exposure to combined hypoxia and hypercapnia, resulting in vasovagal syncope (Halliwill, 2003). Our data of reduced LF/HF (Table 1) are in agreement with that finding. Furthermore, since our subject's ABP was well maintained during hypercapnic exposure in normoxia (Table 1), we speculate that the output of the brain regions responsible for compensatory sympathetic response to hypercapnia might be attenuated during exposure to hypoxia.
An alternative explanation is a blunted peripheral chemoreflex response to severe hypoxia, which might increase an individual's susceptibility to hypoxia-induced syncope due to greater central hypoxic depression associated with greater arterial desaturation. We observed Sp
Implications
The present case report, together with previous findings, highlights the increased risk of syncope for climbers and trekkers upon rapid ascent to high altitude. Accordingly, particular care should be taken during postural changes, whereby impaired compensatory mechanisms, coupled with reduced arterial and cerebral tissue oxygenation associated with hypoxic exposure, would lead to greater risk of hypotension and subsequently greater cerebral hypo-perfusion/tissue deoxygenation during orthostatic challenges.
Conclusions
We present a case of repeated pre-syncope during exposure to relative hypercapnia in a background of severe hypoxia. The development of hypotension suggests an impaired compensatory sympathetic response to an increase in Pa
Footnotes
Acknowledgments
B.K. contributed to the conception and design of the experiment, the interpretation of the data, and the writing of the manuscript. J-L.F. carried out data collection, and led the analysis, interpretation, and writing of the manuscript. Both authors approved the final version of this manuscript.
Author Disclosure Statement
The authors do not declare any competing financial interests.
The Swiss National Science Foundation and the Fondation de Reuter supported this study.