
Abstract
Abstract
MacInnis, Martin J., Nadia Widmer, Utsav Timulsina, Ankita Subedi, Ashmita Siwakoti, Bidur Prasad Pandit, Michael G. Freeman, Eric A. Carter, Irina Manokhina, Ghan Bahadur Thapa, and Michael S. Koehle. A preliminary genome-wide association study of acute mountain sickness susceptibility in a group of Nepalese pilgrims ascending to 4380 m. High Alt Med Biol 16:290–297, 2015.—There is significant interindividual variation in acute mountain sickness (AMS) susceptibility in humans. To identify genes related to AMS susceptibility, we used a genome-wide association study (GWAS) to simultaneously test associations between genetic variants dispersed throughout the genome and the presence and severity of AMS. DNA samples were collected from subjects who ascended rapidly to Gosainkunda, Nepal (4380 m), as part of the 2005, 2010, and 2012 Janai Purnima festivals. The Lake Louise Score was used to measure AMS severity. The primary analysis was based on 99 male subjects (43 with AMS; 56 without AMS). Genotyping for the GWAS was performed using Infinium Human Core Exome Bead Chips (542,556 single-nucleotide polymorphisms were assayed), and validation genotyping was performed with pyrosequencing in two additional cohorts (n = 101 for each). In total, 270,389 single nucleotide polymorphisms (SNPs) passed quality control, and 4 SNPs (one intronic, three nonsynonymous) in the FAM149A gene were associated with AMS severity after correcting for multiple hypothesis testing (p = 1.8E-7); however, in the validation cohorts, FAM149A was not associated with the presence or severity of AMS. No other genes were associated with AMS susceptibility at the genome-wide level. Due to the large influence of environmental factors (i.e., ascent rate and altitude attained) and the difficulties associated with the AMS phenotype (i.e., low repeatability, nonspecific symptoms, potentially independent ailments), we suggest that future studies addressing the variation in the acute human hypoxia response should focus on objective responses to acute hypoxia instead of AMS.
Introduction
A
The relative contributions of genetic and environmental variation to interindividual variation in AMS susceptibility are unclear (MacInnis et al., 2011). Several acute hypoxia-related traits are considered heritable (MacLeod et al., 2013; Masschelein et al., 2014), and genetic variants contribute to acute altitude illness (Kobayashi et al., 2013), chronic altitude illness (Zhou et al., 2013; Wilkins et al., 2014), and high-altitude adaptation (i.e., chronic healthy exposure; Beall et al., 2010; Simonson et al., 2010; Yi et al., 2010); therefore, it is plausible that genetic differences might explain some of the variation in AMS susceptibility.
Multiple genes have been investigated for a role in acute and chronic altitude illnesses [reviewed in MacInnis et al. (2010)]; however, for AMS, none of these associations has been robustly validated. These studies primarily employed candidate gene approaches: genes were investigated because of a priori hypotheses related to their known or supposed physiological functions. Uncertainties about the function of many human genes (Hirschhorn and Daly, 2005) in conjunction with limited knowledge of the physiological mechanisms explaining the variation in AMS susceptibility hinder the candidate gene approach (MacInnis et al., 2011).
In contrast, genome-wide association studies (GWASs) avoid the problem of choosing the correct gene(s) as GWASs do not require a priori hypotheses of the biological mechanisms (Hirschhorn and Daly, 2005). Instead, polymorphisms throughout the genome can be simultaneously interrogated to identify regions that are associated with the phenotype (Hakonarson and Grant, 2011). Through the generation of testable hypotheses, this approach to understanding AMS susceptibility complements a physiological approach.
Recently, genomic approaches have been used to identify genes associated with high-altitude pulmonary edema (HAPE [Kobayashi et al., 2013]), high-altitude pulmonary hypertension (HAPH [Wilkins et al., 2014]), and chronic mountain sickness (Zhou et al., 2013); however, genome-wide approaches have not been applied to AMS, the most common acute altitude illness. The purpose of the study was to determine which (if any) genes were strongly associated with variation in AMS susceptibility. As our sample size was relatively small, we aimed to identify genes that had large effect sizes and to retest these genes in separate samples. We hypothesized that at least some of the variation in AMS susceptibility would be due to genetic variation and that alleles at one or more polymorphism would be over-represented in individuals who developed AMS after ascending to Lake Gosainkunda, Nepal (4380 m).
Methods
Subjects
All subjects attended the Janai Purnima festival at Gosainkunda, Nepal, and they ascended to Gosainkunda (4380 m) from Dhunche (1950 m) in ∼2 days. Subjects were of self-identified Nepalese ancestry. Subject recruitment occurred in Gosainkunda in 2005 (Koehle et al., 2006) and 2010 (MacInnis et al., 2012) and in the town of Dhunche in 2012 (MacInnis et al., 2013a). All subjects were assessed for AMS using the Lake Louise Score (LLS) (Roach et al., 1993) at 4380 m within hours of arrival.
The a priori plan was to identify putative associations using a GWAS and to attempt to replicate these associations in additional samples to reduce the likelihood of false-positive associations. Subjects for the GWAS were selected from the 2010 and 2012 samples, and two validation cohorts were constructed, one entirely collected in 2005 and the other collected in 2010 and 2012. A post hoc decision was made to only analyze the GWAS data of males in the first analysis (see genome-wide genotyping, quality control, and statistical analyses), and the females were added to the second validation cohort.
DNA sample collection and isolation
Saliva was collected in plastic tubes (Saliva DNA Isolation Kit; Norgen Biotek Corp., Thorold, ON, Canada) and cheek cells were scraped from the subjects' inner cheeks with cytobrushes (Fisher Scientific, Ottawa, ON, Canada) and stored in paper envelopes. Samples were transported from Nepal to the University of British Columbia (UBC) for analysis. The manufacturer's protocol was followed to isolate DNA from the saliva samples, and a published protocol was followed to isolate DNA from the cytobrush samples (Saftlas et al., 2004). DNA samples were purified and concentrated using the QIAamp DNA micro kit (Qiagen, Venlo, The Netherlands).
Genome-wide genotyping, quality control, and statistical analyses
DNA from 203 individuals was collected between the 2010 and 2012 Janai Purnima festivals. Many subjects were recruited as families; however, a maximum of one individual per family was used in the analysis (i.e., first, second, and third degree relatives were excluded). To strengthen the case–control analysis, only subjects with an LLS ≥4 and headache score ≥1 (AMS+; n = 73) or with an LLS ≤1 and headache score of 0 (AMS−; n = 71) underwent genome-wide genotyping. One hundred two of these individuals were male and 42 were female.
Genotyping was performed at the Centre for Molecular Medicine and Therapeutics at UBC using Infinium Human Core Exome Bead Chips (Illumina, Inc., San Diego, CA) run on the Illumina 500GX Bead Station. In total, 542,556 polymorphisms, corresponding to an approximately even mix of tagSNPs and exome-focused single nucleotide polymorphisms (SNPs), were interrogated. Thus, genes and intergenic regions were assayed, with a mean coverage of one SNP every 5.5 kb throughout the genome and at least one SNP in ∼19,000 identified genes. Because of a strong association between sex and AMS in this population (Basnyat et al., 2000; MacInnis et al., 2013a), female subjects were excluded from the initial analysis and were instead used for validation of putative associations.
Genotype data were checked for quality using the statistical framework R (r-package.org) and the package, GenABEL (Aulchenko et al., 2007). The entire GWAS sample (n = 144) was used to provide the most robust quality control. A marker was excluded from analysis if (1) its call rate was <98%; (2) its minor allele frequency (MAF) was <1%; or (3) it was not in Hardy–Weinberg Equilibrium (HWE). A subject was excluded from analysis if his/her (1) call rate was <98%; (2) autosomal heterozygosity was too high (False discovery rate = 0.01); or (3) proportion of alleles that were identical by state compared with another sample was ≥0.95. Applying these criteria controlled for low quality DNA samples (call rates), DNA contamination (autosomal heterozygosity), genotyping artifacts (HWE, MAF), and the possibility of duplicated or related samples (identical by state) (Teo, 2009). Following the recommendations from Teo (2009), only those markers and individuals who passed quality control were analyzed.
Analysis of the genome-wide data was performed using the GenABEL package in R (Aulchenko et al., 2007). For the primary analysis, polymorphisms were tested for associations with the presence/absence of AMS (i.e., AMS was treated as a binary trait): male subjects were divided into AMS+ and AMS− groups, and polymorphisms were tested for association with AMS status using χ2 tests. A second analysis was performed in which polymorphisms were tested for associations with the severity of AMS (i.e., AMS was treated as a quantitative trait). For both analyses, age was used as a covariate.
Lambda, which (when greater than 1) signals the presence of population stratification, was <1 (0.97) for both analyses. As a result, it was not necessary to employ a method of genomic control in either analysis. The possibility of genetic substructure being present in the dataset was explored further using classical multidimensional scaling to plot and group subjects based on their genetic relatedness with each other (i.e., kinship). For all genome-wide analyses, associations were deemed to be genome-wide significant at a 5% empirical significance cutoff that was determined through 200 permutations implemented in GenABEL. We had adequate power (0.78) to detect large effect sizes (cohen's w = 0.5) after correcting for multiple hypotheses (alpha = 1.8E-7).
Validation stage
The a priori plan was to retest putative associations with samples collected in 2005 (n = 101) and the remaining samples from 2010 to 2012 (n = 59). As already discussed, a post hoc decision was made to add the female subjects who were genotyped using the Human Core Exome Bead Chip (n = 42) to the 2010/2012 validation group for a sample size of n = 101.
Based on the results of the male-only GWAS, the validation samples were assessed for the nonsynonymous FAM149A variant c.121A>G (rs4862650) using a pyrosequencing-based genotyping assay run on a PyroMark Q24 (Qiagen, Inc., Toronto, ON, Canada). The genotyping assay was designed with the PSQ Assay Design software (Biotage, Uppsala, Sweden), and primer sequences were as follows: For: 5′-TCTCTTTGACCTCGCAGAGTATTA-3′, Rev: 5′-biotin-TCTCTGATGTTGCTGCTGTGT-3′, sequencing primer: 5′-ACAGCTGATCCTGCCC-3′. The 15-μL polymerase chain reaction (PCR) mixture contained 1× PCR buffer, 1.5 nM MgCl2, 0.2 nM dNTPs, 0.4 nM primers, 1 U Platinum Taq polymerase (Invitrogen, Carlsbad, CA), and 100 ng DNA template. PCR conditions were as follows: initial denaturation for 4 minutes, followed by 40 cycles at 95°C for 30 seconds, 59°C for 30 seconds, and 72°C for 30 seconds, with a final elongation step at 72°C for 4 minutes. The PCR product was then washed and hybridized according to the manufacturer's instructions and genotyped using a PyroMark Q24 protocol.
In the validation cohorts, χ2 tests were used to compare the frequency of genotypes between AMS+ and AMS− individuals, and a Mann–Whitney U-test was used to compare the severity of AMS across genotypes. Associations in the validation cohorts were considered statistically significant at p < 0.05.
Results
Genotype data quality control
In total, 265,063 SNPs were removed predominately because of violations of the MAF but also due to violations of HWE and genotyping call rate requirements; 13,441 genes had at least one SNP assayed. Three male subjects were removed from the analysis because of unsatisfactory genotyping call rates. Subject characteristics for the remaining 99 subjects are presented in Table 1. When subjects were grouped into clusters based on multidimensional scaling (k = 2, 3, and 4 clusters), the incidence of AMS did not differ between clusters (data not shown). The AMS+ group was significantly older for all datasets (Table 1).
Data for age and LLS are presented as means and standard deviations.
Male cohort following quality control.
AMS+, positive diagnosis for acute mountain sickness; AMS−, negative diagnosis for acute mountain sickness; LLS, Lake Louise Score; NA, not applicable; GWAS, genome-wide association study.
Genome-wide analysis: AMS+ versus AMS−
The Manhattan plot showing the results of the χ2 tests (i.e., treating AMS as a binary trait) is presented in Figure 1A. None of the polymorphisms reached genome-wide statistical significance. The 20 SNPs with the smallest p values are reported in Table 2.
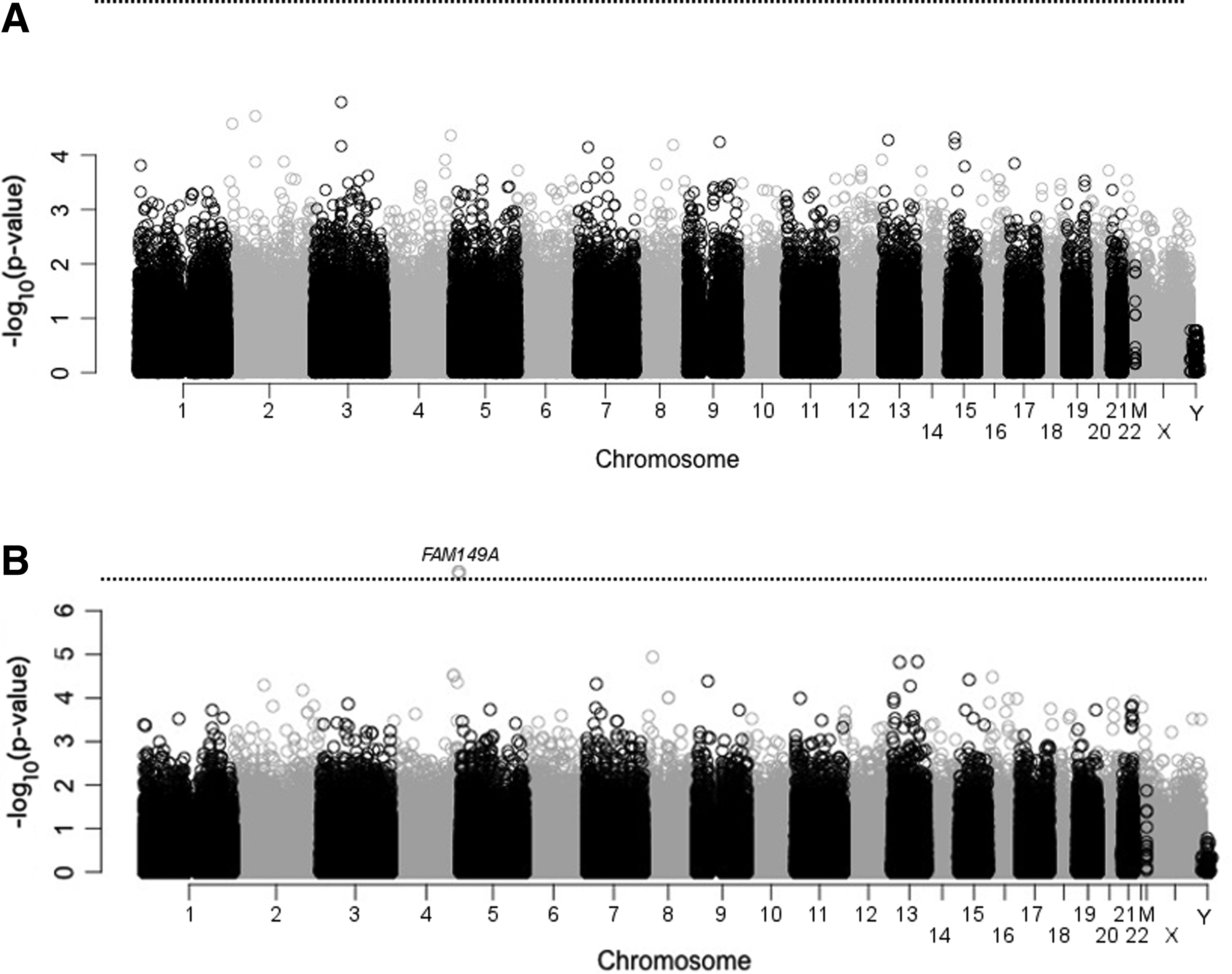
Manhattan plots of the −log(p values) for statistical tests of
Based on assembly GRCh37.p13, annotation release 105.
p-Values were corrected using 200 permutations implemented in GenABEL.
Genome-wide analysis: AMS severity
The Manhattan plot showing the results of the linear regression tests between polymorphisms and LLS is presented in Figure 1B. Four SNPs in the FAM149A gene reached genome-wide statistical significance, including one intronic SNP (rs7684126) and three nonsynonymous SNPs (rs4862650, rs4862653, rs2276924). Summary information for the 20 SNPs with the smallest p values is reported in Table 3. The four associated SNPs were in linkage disequilibrium. The genotypes associated with more severe AMS were G/A, Glu/Lys, Glu/Lys, and Arg/His and the genotypes associated with less severe AMS were G/G, Glu/Glu, Glu/Glu, and Arg/Arg (genotypes are listed in the order in which rs numbers are presented above). The mean LLSs for male subjects with these genotypes are shown in Figure 2.

The mean LLS for the genome-wide association study (GWAS) and validation cohorts partitioned by the rs4862650 genotype. The asterisk (*) denotes that the mean LLS was statistically greater for the A/A+A/G genotypes (A−) versus the G/G genotype in the GWAS sample at the genome-wide level. The error bars represent the standard error of the mean.
Based on assembly GRCh37.p13, annotation release 105.
p-Values were corrected using 200 permutations implemented in GenABEL.
SNPs, single nucleotide polymorphisms.
Validation of FAM149A association
Due to the linkage disequilibrium among associated SNPs, only one of the four SNPs (rs4862650) was chosen for further testing in the validation samples. Neither the genotype nor allele frequency was associated with AMS status in either validation cohort (Table 4). Similarly, AMS severity was not associated with the genotype from this SNP (Fig. 2). Excluding females from the validation cohorts did not alter the results (data not shown).
HWE, Hardy–Weinberg Equilibrium.
Discussion
This is the first GWAS of AMS susceptibility. Four linked polymorphisms (one intronic SNP and three nonsynonymous SNPs) located in the FAM149A gene were associated with AMS severity in the initial GWAS; however, the SNP chosen for retesting was not associated with AMS in the two validation cohorts. Therefore, it is possible that this putative association was a false positive. No other genes, including those previously associated with acute altitude illness susceptibility [see MacInnis et al. (2011) for a review], were associated with AMS in this study (at genome-wide statistical significance); however, due to the small sample size (and therefore relatively low statistical power for small effect sizes), this study cannot rule out the possibility that other polymorphisms/genes with small effect sizes are associated with AMS.
The four polymorphisms putatively associated with AMS severity in this study were all within ∼6500 bp and were tightly linked. Three of the polymorphisms were in the coding region of the FAM149A gene and led to amino acid substitutions, and the other polymorphism was an intron variant. Three simultaneous amino acid substitutions might have an effect on the FAM149A protein, but it is difficult to speculate what the effect might be given that the function of the protein is unknown. Alternatively, the FAM149A gene is located on chromosome 4 near several other genes: TLR3, CYP4V2, KLKB1, and F11.
Although the association between FAM149A and AMS was not confirmed in the validation samples, FAM149A is an interesting candidate for AMS susceptibility: the expression of FAM149A is highest in the trigeminal ganglion and superior cervical ganglion (Su et al., 2004). The former region is central in two proposed models of AMS pathophysiology (Sanchez del Rio and Moskowitz, 1999; Hackett and Roach, 2001; Bartsch and Bailey, 2013), and the latter region innervates numerous organs and tissues in the head and trunk, including cerebral blood vessels (Mitchell et al., 2009), and is involved in the regulation and protection of cerebral circulation (Treggiari et al., 2003; Cassaglia et al., 2008). While the lack of replication suggests that FAM149A was a false positive, the plausible role of these tissues in monitoring and regulating cerebral circulation at least makes FAM149A a candidate for a role in AMS susceptibility.
AMS is a complex condition to study from a genetic perspective. First, AMS develops (over several hours) in response to environmental stimuli that mostly occur in remote locations. Accordingly, collecting a large number of samples is difficult. Furthermore, the rate of ascent (Bloch et al., 2009) and the altitude attained (Maggiorini et al., 1990) appear to be the most important factors affecting the incidence of AMS and they are difficult to control within and between studies. It is possible that even if AMS susceptibility is genetically influenced, the ascent rate and altitude are more important in determining susceptibility. Alternatively, the repeatability of AMS appears to be low (MacInnis et al., 2014, 2015)—especially relative to a condition such as HAPE (Bartsch et al., 2002)—meaning that it might be necessary to assess an individual's susceptibility on multiple occasions before assigning a phenotype. Many subjects in our sample were altitude naïve, which could affect the accuracy of the phenotype as well (MacInnis et al., 2014). Finally, the current questionnaire used to measure AMS also appears to measure multiple clinical syndromes (MacInnis et al., 2013b; Hall et al., 2014), which is problematic if certain genes influence particular responses to hypoxia (i.e., sleep vs. headache).
For similar reasons, replicating associations between specific genes and AMS susceptibility is also difficult. None of the genes previously associated with AMS (see MacInnis et al., 2010) were replicated in this study. Those previous associations could be false positives, but it is also possible that (1) our study was underpowered to detect the effect sizes of those variants, (2) the polymorphisms are only associated with AMS in specific biogeographical populations, (3) the polymorphisms are only associated with AMS for specific ascent conditions, or (4) the associated polymorphisms (and any linked polymorphisms) were not assayed. For the latter point, many SNPs were not analyzed in the present study because of a lower MAF. This is likely a result of our chip not being optimized for individuals of Nepalese ancestry.
Although AMS is the phenotype of interest from a public health perspective, investigating objective measures of the acute hypoxic response [e.g., decline in VO2max (Masschelein et al., 2015)] might be a more fruitful approach to gain insights into the influence of genetics on responses to acute hypoxia. In addition to the advantages gained from more objective measurements of hypoxia tolerance in humans, because susceptibility to AMS is likely multifactorial in nature, a better understanding of acclimatization might be realized when responses to hypoxia are studied at the system level (i.e., a more reductionist approach).
Aside from issues related to the AMS phenotype, there are several limitations to our analysis that must be appreciated when interpreting our results. First, our sample size was limited by the cost of the genotyping. GWASs normally require sample sizes >1000 subjects to overcome the necessary corrections for multiple hypothesis testing; therefore, small sample sizes normally prevent any SNPs from reaching genome-wide statistical significance. Knowing this before the analysis began, we planned to (1) retest putative associations in an independent sample and (2) use our data as an exploratory study from which candidate genes could be chosen for future experiments. Second, a post hoc decision was made to remove the female subjects from the first round of analysis. There was a marked difference in the incidence of AMS between sexes (43% in males vs. 70% in females) that we hypothesized was linked to cultural differences in the reporting of symptom severities (Basnyat et al., 2000; MacInnis et al., 2013a). When the FAM149A gene was tested for associations with AMS in the female cohort, the association was not statistically significant. Finally, whether or not this study can be generalized to other ascent profiles and durations of hypoxic exposure is unknown.
Conclusion
This is the first GWAS of AMS. Four SNPs in the FAM149A gene reached genome-wide statistical significance for an association with AMS severity in the first round of analysis; however, this gene was not associated with AMS in the validation sample. The function of this gene is unknown; however, previous studies demonstrated that its expression was markedly greater in the trigeminal and superior cervical ganglia, which are sites plausibly involved in acute AMS pathophysiology. Testing this gene for an association with AMS in other populations is warranted. As AMS cannot be measured objectively and because an individual's AMS susceptibility is difficult to ascertain on one ascent, we recommend that future studies should focus on specific symptoms of AMS (e.g., headache) or objective markers of acute hypoxia tolerance other than AMS susceptibility.
Footnotes
Acknowledgments
The Natural Sciences and Engineering Research Council of Canada (NSERC) provided research funding for this project. The authors thank the Nepal Health Research Council for guidance on the study design, and the district health, public health, and chief district officers of Rasuwa for permitting data collection in Dhunche and Gosainkunda. The authors are also thankful for logistical advice and assistance from members of the Mountain Medicine Society of Nepal and members of the Himalayan Rescue Association, in particular Dr. Ashish Lohani, Bhuwan Acharya, and Gobi Bashyal. The kind staff at the Langtang View Hotel (Dhunche), the Hotel Lakeside (Gosainkunda), and various local businesses in Dhunche assisted with data collection by providing space and food to subjects and researchers. Finally, the authors are grateful to the many Nepalese pilgrims who took time out of their pilgrimage to participate in the study.
Author Disclosure Statement
No competing financial interests exist.