
Abstract
Abstract
Junming Fan, Xiaofang Fan, Yang Li, Lu Ding, Qingqing Zheng, Jinbin Guo, Dongmei Xia, Feng Xue, Yongyu Wang, Shufang Liu, and Yongsheng Gong. Chronic normobaric hypoxia induces pulmonary hypertension in rats: role of NF-κB. High Alt Med Biol 17:43–49, 2016.—To investigate whether nuclear factor-kappa B (NF-κB) activation is involved in chronic normobaric hypoxia-induced pulmonary hypertension (PH), rats were treated with saline or an NF-κB inhibitor, pyrrolidine dithiocarbamate (PDTC, 150 mg/kg, sc, twice daily), and exposed to normoxia or chronic normobaric hypoxia with a fraction of inspired oxygen of ∼0.1 for 14 days. Lung tissue levels of NF-κB activity, and interleukin (IL)-1β, IL-6, and cyclooxygenase-2 mRNAs, were determined, and mean pulmonary arterial pressure, right ventricular hypertrophy, and right heart function were evaluated. Compared to the normoxia exposure group, rats exposed to chronic normobaric hypoxia showed an increased NF-κB activity, measured by increased nuclear translocation of p50 and p65 proteins, an increased inflammatory gene expression in the lungs, elevated mean pulmonary arterial blood pressure and mean right ventricular pressure, right ventricular hypertrophy, as assessed by right ventricle-to-left ventricle plus septum weight ratio, and right heart dysfunction. Treatment of hypoxia-exposed rats with PDTC inhibited NF-κB activity, decreased pulmonary arterial blood pressure and right ventricular pressure, and ameliorated right ventricular hypertrophy and right heart dysfunction. Hypoxia exposure increased protein kinase C activity and promoted pulmonary artery smooth muscle cell proliferation in vitro. Our data suggest that NF-κB activation may contribute to chronic normobaric hypoxia-induced PH.
Introduction
P
Nuclear factor-kappa B (NF-κB) is a proinflammatory transcription factor that mediates inflammatory responses by inducing the expression of large numbers of proinflammatory genes. The NF-κB family of proteins consists of 5 members: p65 (RelA), p50 (NF-κB1), p52 (NF-κB2), c-Rel, and RelB. Under physiological conditions, NF-κB dimer is sequestered in the cytoplasm by its inhibitory protein, IκB. Under pathological conditions, cell stress or inflammatory stimuli activate IκB kinase, which phosphorylates IκBα, leading to ubiquitination by SCFFWD1-βTrCP ubiquitin ligase and subsequent proteasomal degradation of IκBα protein. The released NF-κB dimer translocates to the nucleus, where it induces transcription of NF-κB target genes (Siebenlist et al., 1994; Gilmore, 2006; Oeckinghaus et al., 2011). Pyrrolidine dithiocarbamate (PDTC) inhibits NF-κB activation by repressing the ubiquitin ligase activity and inhibiting IκB ubiquitination and degradation (Liu et al., 1999; Hayakawa et al., 2003). NF-κB plays roles in many pathological processes, including monocrotaline-induced PH (Sawada et al., 2007; Huang et al., 2008; Kimura et al., 2009; Hosokawa et al., 2013; Lu et al., 2013; Price et al., 2013; Wang et al., 2013; Li et al., 2014). However, the role of NF-κB in chronic normobaric hypoxic PH has not been studied.
Hypoxia occurs under physiological conditions, such as in people living at high altitude (Bahrke and Shukitt-Hale, 1993; Bartsch and Swenson, 2013; Netzer et al., 2013; Welsh and Peacock, 2013), and pathological conditions, such as in patients with chronic obstructive pulmonary disease and sleep apnea syndrome (Naeije and Vanderpool, 2013; Scherrer et al., 2013; Welsh and Peacock, 2013). Chronic hypoxia is a major factor for the induction of PH, although the mechanisms underlying hypoxia-induced PH are not fully understood. Most of the previous studies exposed animals to hypobaric hypoxia. There have been few studies examining the effects of chronic normobaric hypoxia (Yang et al., 2012), an experimental condition that is more clinically relevant.
The aims of this study were to evaluate the effect of chronic normobaric hypoxia on the development of PH in rats and investigate whether NF-κB activation is involved in the pathological process. Our data suggest that NF-κB activation may contribute to the development of chronic normobaric hypoxia-induced PH.
Materials and Methods
Animals
Male Sprague Dawley rats weighing 250 ± 20 g, provided by the Animal Center of Wenzhou Medical University (Wenzhou, Zhejiang, China), were maintained at 22°C ± 1°C and relative humidity of 50% ± 5% under a 12-hour light–12-hour dark cycle with food and water ad libitum. Rats were allowed to acclimate to the animal facility for at least 1 week. All animal study protocols were approved by the Institutional Animal Care and Use Committee of the Wenzhou Medical University and complied with the regulations of the Ministry of Health, China, and the U.S. National Institutes of Health Guidelines for the use and care of laboratory animals. All efforts were made to minimize animal suffering and reduce number of animal use.
Hypoxia exposure
Rats were randomly divided into normoxia and hypoxia groups (n = 8 per group). For hypoxia exposure, rats were placed in a normobaric hypoxia exposure chamber with a fraction of inspired oxygen (FIO2) of ∼0.1, 23 hours per day, for 2 weeks. Rats in the normoxia group were placed in a chamber with FIO2 of ∼0.21. Cages were cleaned daily.
PDTC treatment
We studied four groups (eight per group) of rats: normoxia+vehicle, normoxia+PDTC, hypoxia+vehicle, and hypoxia+PDTC. Rats in the normoxia+vehicle and hypoxia+vehicle groups were subcutaneously injected with 200 μL saline, and those in the normoxia+PDTC and hypoxia+PDTC groups were subcutaneously injected with PDTC (150 mg/kg in 200 μL saline), twice daily, and exposed to normoxia or normobaric hypoxia, as described above. PDTC was freshly prepared in normal saline (pH 7.4) each day before injection.
Measurements of pulmonary arterial pressure and right ventricular hemodynamics
After the final hypoxia exposure, mean pulmonary arterial pressure (mPAP) and right ventricular hemodynamics were measured, as we previously described (Mao et al., 2014). In brief, rats were anesthetized and ventilated. A microcatheter was inserted through jugular vein into right ventricle (RV) and then pulmonary artery. After a 30-minute equilibration period, right ventricular pressure, right ventricular end-systolic pressure (RVESP), right ventricular end-diastolic pressure (RVEDP), maximal rate of RV systolic pressure development (RV +dp/dt max), maximal rate of RV diastolic pressure development (RV dp/dt min), and mPAP were recorded on a physiological recorder (PowerLab) via a transducer (PowerLab 8 passages electrophysiolograph; ADI) connected to the microcatheter.
Assessment of right ventricular hypertrophy
After mPAP measurement, rats were sacrificed, and the hearts were collected. Atrium was trimmed, and the free wall of the RV was separated from the left ventricle (LV). RV and LV+septum (LV+S) of each heart were weighted, and RV/LV+S ratio was calculated to assess right ventricular hypertrophy.
Assessment of pulmonary artery smooth muscle cell proliferation in vitro
Rat pulmonary artery smooth muscle cells (PASMCs) were isolated, as previously described (Xia et al., 2011; Mao et al., 2014). Cells were cultured in DMEM supplemented with 10% fetal calf serum, 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 0.25 mg/mL amphotericin B. PASMC phenotype was confirmed by cell morphology and α-smooth muscle actin (α-SMA)-positive staining. PASMCs (three to five passages, 2.5 × 105) were incubated in 10% CCK-8 reagent (Dojindo) diluted in culture media at 37°C and exposed to 21% O2 and 5% CO2, for the normoxia group, or 1% O2 and 5% CO2, for the hypoxia group, in an incubator (Thermo). Proliferation rates were determined following 24-hour hypoxia exposure, according to the manufacturer's protocol.
Quantitative real-time polymerase chain reaction
Total RNAs were isolated from the lungs using QIAGEN RNeasy Kit, and the first-strand cDNA was synthesized with an RNA PCR Kit (QIAGEN) following the manufacturer's instructions. Quantitative real-time polymerase chain reaction (PCR) was performed in an ABI PCR System using RT2 Profiler PCR Kit and RT2 SYBR Green qPCR Mastermixes (QIAGEN). The β-actin gene was used as an internal control. The primer sequences used for PCR were shown in Table 1. Reactions were performed following the manufacturer's protocol. For each gene, the reactions were performed in triplicate, and relative gene expression level was normalized to β-actin gene expression. Data were analyzed by the 2−ΔΔCt method.
COX-2, cyclooxygenase-2; IL, interleukin.
Western blot
The lungs were lysed in an RIPA buffer (1% Triton X-100, 0.5% sodium deoxycholate, 0.2% SDS, 150 mM NaCl, 10 mM HEPES, pH 7.3, 2 mM EDTA, and protease inhibitor mixture; Pierce). The lysates were homogenized on ice by passing 20 times through a 22-gauge needle. The cytosolic and nuclear proteins were prepared from the whole-lung homogenates using cytosolic and nuclear protein extraction kit (Applygen), according to the manufacturer's protocol. Protein concentration was measured using the BCA Protein Assay Kit (Pierce). Fifty micrograms of protein was loaded, separated on a 10% SDS–PAGE, and transferred to a nitrocellulose membrane. The membrane was blocked in 5% nonfat dried milk in PBS and 0.1% Tween 20 and incubated with primary antibodies overnight at 4°C, followed by incubation with appropriate secondary antibodies for 1 hour at room temperature. The primary antibodies used include p50, p65, phosphorylated extracellular signal-regulated kinases (p-ERKs), phosphorylated protein kinase C (p-PKC), GAPDH, and lamin B (Table 2). After washing, the protein bands were visualized by ECL Western Blotting Detection System (Amersham Biosciences). The Western blot protein bands were quantified using densitometry, analyzed using ImageJ System, and expressed as a percentage of control.
α-SMA, α-smooth muscle actin; p-ERK, phosphorylated extracellular signal-regulated kinase; p-PKC, phosphorylated protein kinase C.
Statistical analysis
Data were expressed as the mean ± standard deviation. Statistical analysis was carried out using the Prism software package (GraphPad 5.0). The differences between two groups were analyzed using unpaired Student's t-tests. One-way ANOVA was performed if more than two groups were compared. A p-value <0.05 was considered significant.
Results
Chronic normobaric hypoxia induces PH
Rats exposed to normobaric hypoxia for 2 weeks had an elevated mPAP compared to those exposed to normoxia (Fig. 1A). In addition, normobaric hypoxia-exposed rats displayed an increased mean right ventricular pressure (Fig. 1B) and increased RV-to-LV+S weight ratio (Fig. 1C). The morphological changes were associated with functional changes. Hypoxia-exposed rats showed significantly increased RVESP (Fig. 1D), RVEDP (Fig. 1E), and maximal rate of RV systolic pressure development (RV +dp/dt max) (Fig. 1F) and a decreased rate of RV diastolic pressure development (RV −dp/dt min) (Fig. 1G). These results indicated that chronic normobaric hypoxia exposure induces PH, right ventricular hypertrophy, and right heart dysfunction.

Chronic normobaric hypoxia induces pulmonary hypertension (PH), right ventricular hypertrophy, and right ventricular dysfunction. Rats were exposed to normobaric hypoxia for 14 days. Mean pulmonary arterial pressure (mPAP) and right heart hemodynamics were recorded by right heart and pulmonary artery catheterization, followed by collection of the heart for determining right ventricle-to-left ventricle plus septum weight ratio [RV/(LV+S)]. Hypoxia exposure increased mPAP
To determine whether chronic normobaric hypoxia-induced PH is associated with inflammation, we compared lung tissue levels of interleukin (IL)-1β, IL-6, and cyclooxygenase-2 (COX-2) mRNA expressions between normoxia- and hypoxia-exposed rats. We found that rats with hypoxic PH had significantly increased lung tissue levels of IL-1β mRNA and COX-2 mRNA (Fig. 2), indicating that development of hypoxic PH is associated with inflammation following chronic normobaric hypoxia exposure.
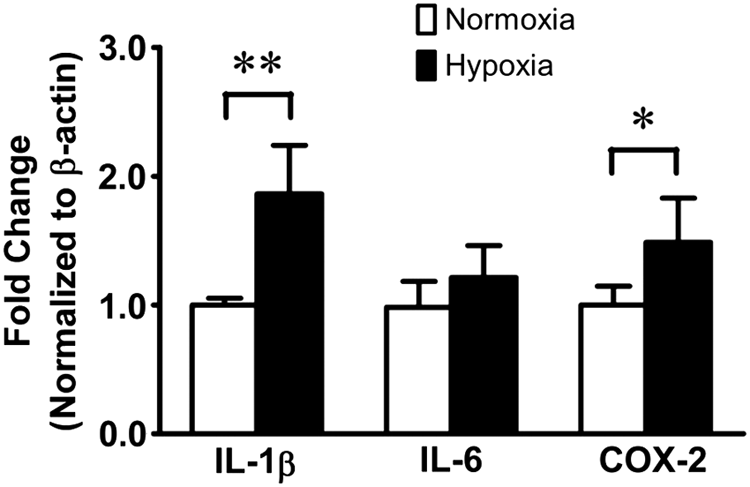
Chronic normobaric hypoxia induces inflammatory gene expression in the lungs. Rats were exposed to normoxia or chronic normobaric hypoxia for 14 days, and lung tissue levels of interleukin (IL)-1β, IL-6, and cyclooxygenase-2 (COX-2) mRNA expressions were quantified by real-time polymerase chain reaction. All data are shown as mean ± SD. *p < 0.05, **p < 0.01 versus normoxia; n = 8 per group.
Inhibition of NF-κB activation alleviates normobaric hypoxia-induced PH
To examine the role of NF-κB in chronic normobaric hypoxia-induced PH, we treated rats with PDTC, an inhibitor of NF-κB activation, and examined the effects of PDTC on normobaric hypoxia-induced PH, right ventricular hypertrophy, and right ventricular dysfunction.
Compared with normoxia-exposed rats, rats exposed to hypoxia showed significantly increased mPAP, mRVP, RV/(LV+S), RVESP, RVEDP, and RV +dp/dt max and a decreased RV−dp/dt min (Fig. 3). Treatment of hypoxia-exposed rats with PDTC reversed hypoxia-induced increases in mPAP, mRVP, RV/(LV+S), RVESP, and RVEDP and tended to reverse hypoxia-induced increase in RV +dp/dt max and decrease in RV−dp/dt min (Fig. 3).
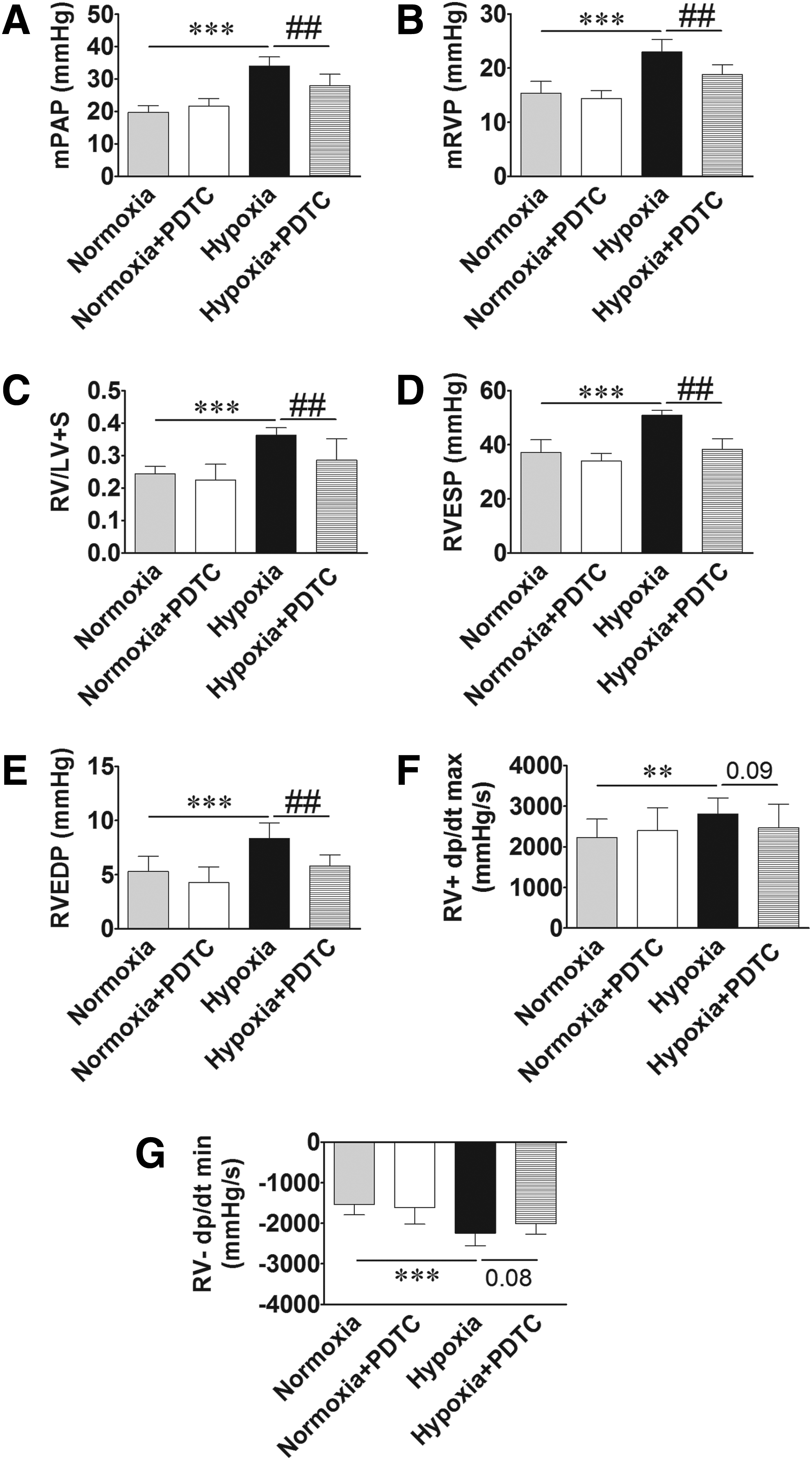
Inhibition of nuclear factor-kappa B (NF-κB) activation by pyrrolidine dithiocarbamate (PDTC) ameliorates chronic normobaric hypoxia-induced PH, right ventricular hypertrophy, and right ventricular dysfunction. Rats were treated with saline or PDTC (an NF-κB inhibitor) and exposed to normoxia or chronic normobaric hypoxia for 14 days. mPAP and right heart hemodynamics were recorded as described above. PDTC treatment mitigated hypoxia-induced increase in mPAP
We confirmed that PDTC inhibits NF-κB activation in our model by showing that PDTC prevented hypoxia-induced nuclear translocation of p50 and p65 proteins. Western blot demonstrated that hypoxia-exposed rats had markedly increased nuclear levels and decreased cytoplasmic levels of NF-κB p50 and p65 proteins in the lungs (Fig. 4), suggesting that hypoxia promotes NF-κB p50 and p65 nuclear translocation. Treatment of hypoxia-exposed rats with PDTC attenuated the hypoxia-induced increase or decrease in nuclear or cytoplasmic levels of p50 and p65 proteins (Fig. 4), suggesting that PDTC inhibits p50 and p65 nuclear translocation. Taken together, these results suggest that NF-κB activation may contribute to chronic normobaric hypoxia-induced PH, right ventricular hypertrophy, and right heart dysfunction.
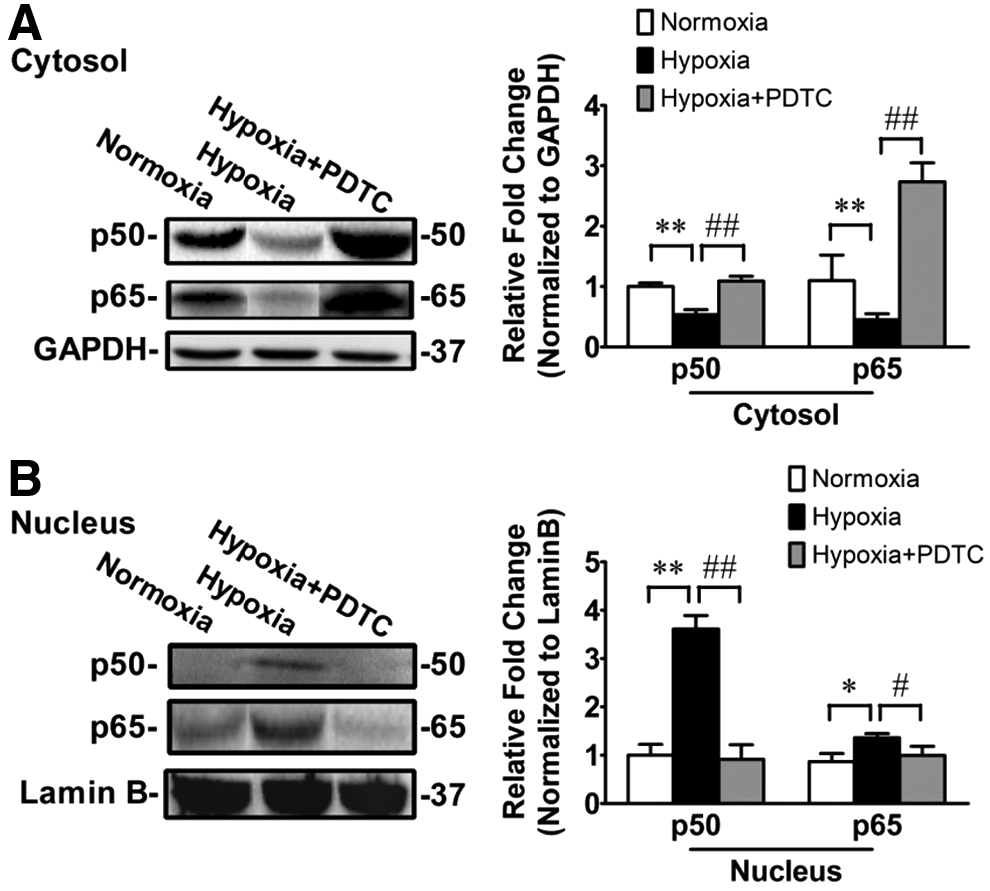
PDTC inhibits chronic normobaric hypoxia-induced NF-κB activation. Rats were treated with saline or PDTC and exposed to normoxia or chronic normobaric hypoxia for 14 days. Lung tissue level of NF-κB activity was determined by measuring p50 and p65 nuclear translocation using Western blot. Levels of p50 and p65 proteins significantly decreased in cytoplasmic fraction
Hypoxia stimulates PASMC proliferation in vitro
To examine whether PASMC proliferation plays a role in hypoxia-induced PH, primary PASMCs were isolated and cultured, and phenotype was confirmed by cell morphology and staining with vascular smooth muscle cell-specific marker, α-smooth muscle action (α-SMA), antibody (Fig. 5A). PASMCs were exposed to normoxia or hypoxia for 24 hours, and proliferation rate was assessed using CCK-8 assay kit. Hypoxia exposure increased PASMC proliferation rate (Fig. 5D). Hypoxia exposure increased p-PKC but did not increase p-ERKs in PASMCs, suggesting that hypoxia may promote PASMC proliferation by activating PKC but not mitogen-activated kinase in PASMCs.
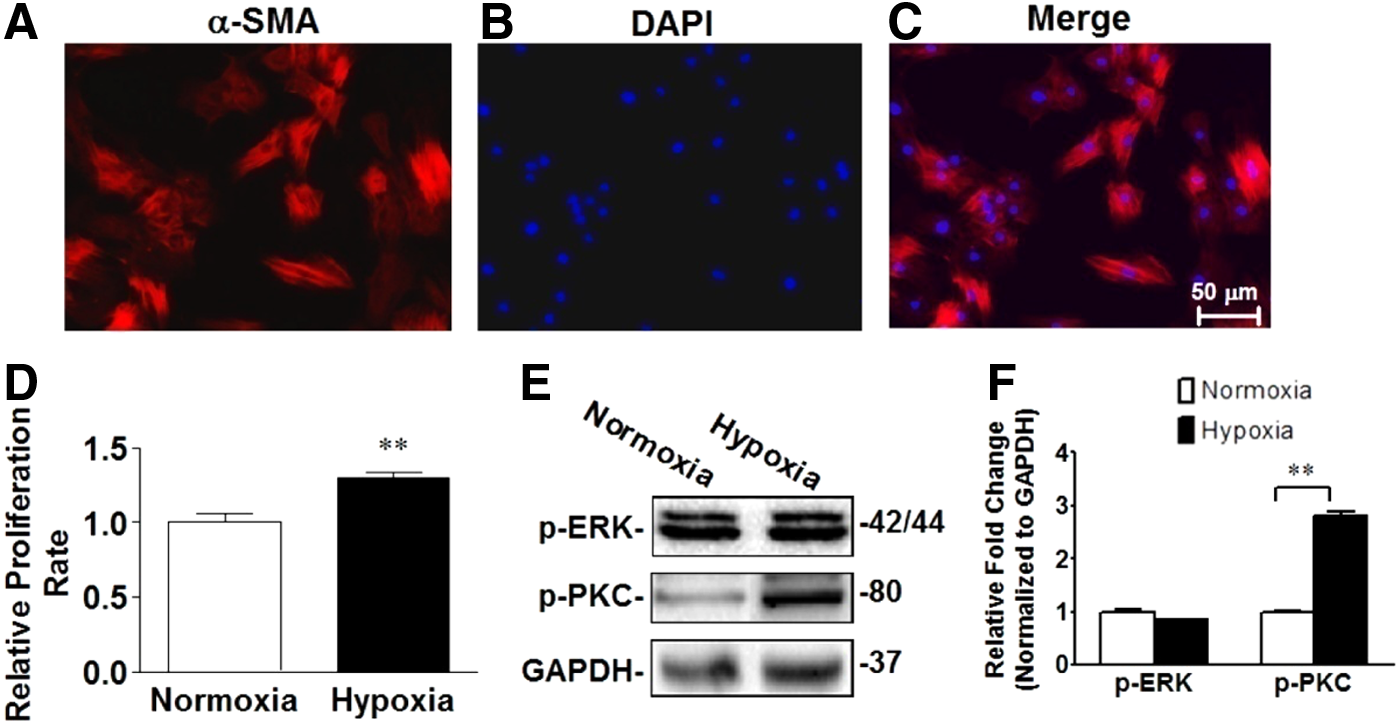
Hypoxia promotes pulmonary artery smooth muscle cell (PASMC) proliferation in vitro.
Discussion
This study demonstrated that rats exposed to chronic normobaric hypoxia developed PH, right ventricular hypertrophy, and right heart dysfunction. More importantly, we demonstrated that chronic normobaric hypoxia-induced PH may involve the NF-κB pathway. Development of PH following chronic normobaric hypoxia exposure was associated with a markedly increased NF-κB activity and increased expression of NF-κB target genes in the lungs. More importantly, treatment of hypoxia-exposed rats with an NF-κB inhibitor, PDTC, suppressed NF-κB activity and ameliorated normobaric hypoxia-induced PH and right ventricular hypertrophy.
Exposure of animals to chronic hypoxia has been used for decades to study the mechanisms of hypoxia-induced pathologies both in altitude (hypobaric hypoxia) sickness and in chronic obstructive pulmonary disease (normobaric hypoxia)-associated hypoxic PH (Stenmark et al., 2006; Sajkov and McEvoy, 2009; Weitzenblum et al., 2009; Yang et al., 2012). However, only one study has examined the effect of normobaric hypoxia. Yang et al. (2012) reported that chronic normobaric hypoxia exposure induced right ventricular hypertrophy in mice. The prior study did not measure mPAP. Our current study extends the previous study by showing that chronic normobaric hypoxia exposure caused an increased mPAP and right ventricular hypertrophy.
Many previous studies have demonstrated that PH is linked to inflammation (Dolenc et al., 2014; Groth et al., 2014; Hashimoto-Kataoka et al., 2015; Matura et al., 2015; Pugliese et al., 2015). Clinical studies showed that NF-κB is activated in pulmonary endothelial cells, PASMCs, and immune cells in patients with idiopathic pulmonary arterial hypertension (PAH) (Price et al., 2013) and that patients with idiopathic PAH exhibited elevated serum levels of inflammatory cytokines, which may correlate with a higher mortality and worse outcomes (Soon et al., 2010). Animal studies demonstrated that chronic hypoxia exposure activated NF-κB in pulmonary vessels and the lungs, and inhibition of NF-κB activation ameliorated hypobaric hypoxia- or monocrotaline-induced PH, pulmonary vascular remodeling, and right ventricular hypertrophy (Sawada et al., 2007; Huang et al., 2008; Kimura et al., 2009; Hosokawa et al., 2013; Price et al., 2013; Li et al., 2014). Consistent with these reports, proinflammatory cytokine was found to exaggerate hypoxia-induced PH (Steiner et al., 2009), whereas anti-inflammatory cytokine, IL-10, attenuates monocrotaline-induced PH (Ito et al., 2007). These findings support the view that activation of inflammatory pathways plays an important role in the pathogenesis of PH.
We demonstrated that chronic normobaric hypoxia exposure activated NF-κB and induced inflammatory cytokine expression in the lungs, and inhibition of NF-κB ameliorated the development of PH, right ventricular hypertrophy, and right ventricular dysfunction. Our results are consistent with, but extend, previous studies (Sawada et al., 2007; Huang et al., 2008; Kimura et al., 2009; Hosokawa et al., 2013; Price et al., 2013; Li et al., 2014) by showing that inflammation is also an important mechanism of chronic normobaric hypoxia-induced PH.
Chronic hypoxia stimulates PASMC proliferation, which contributes to pulmonary vascular remodeling (Davie et al., 2004; Stenmark et al., 2006) and the progression of PH (Morrell et al., 2009). Activation of PKC or ERK1/2 pathway participates in the regulation of PASMC proliferation (Zhang et al., 1998; Agbani et al., 2011; Lu et al., 2013). Consistent with those reports, our current results showed that chronic hypoxia promoted PASMC proliferation with activation of PKC but not ERK pathway. Further studies are needed to clarify the role of PKC in hypoxia-induced PASMC proliferation.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81200039), the Major State Basic Research Development Program of China (2012CB518200), the Natural Science Foundation of Zhejiang Province of China (LY12H01004 and LY16H010006), the Foundation of Zhejiang Educational Committee (Y201430622), and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry.
Author Disclosure Statement
The authors declare no conflicts of interest associated with this article.