
Abstract
Abstract
MacInnis, Martin J., and Michael S. Koehle. Evidence for and against genetic predispositions to acute and chronic altitude illnesses. High Alt Med Biol. 17:281–293, 2016.—Humans exhibit marked variation in their responses to hypoxia, with susceptibility to acute and chronic altitude illnesses being a prominent and medically important example. Many have hypothesized that genetic differences are the cause of these variable responses to hypoxia; however, until recently, these hypotheses were based primarily on small (and sometimes anecdotal) reports pertaining to apparent differences in altitude illness susceptibility between populations, the notion that a history of altitude illness is indicative of subsequent risk, the heritability of hypoxia-related traits, and candidate gene association studies. In the past 5 years, the use of genomic techniques has helped bolster the claim that susceptibility to some altitude illnesses is likely the result of genetic variation. For each of the major altitude illnesses, we summarize and evaluate the evidence stemming from three important characteristics of a genetic trait: (1) individual susceptibility and repeatability across assessments, (2) biogeographical differences and familial aggregation, and (3) association(s) with genetic variants. Evidence to support a genetic basis for susceptibilities to acute mountain sickness (AMS) and high-altitude cerebral edema (HACE) is limited, owing partially to the subjective and unclear phenotype of AMS and the rarity and severity of HACE. In contrast, recent genomic studies have identified genes that influence susceptibility to high-altitude pulmonary edema, chronic mountain sickness, and high-altitude pulmonary hypertension. The collection of more individual, familial, and biogeographical susceptibility data should improve our understanding of the extent to which genetic variation contributes to altitude illness susceptibility, and genomic and molecular investigations have the potential to elucidate the mechanisms that underpin altitude illness susceptibility.
Introduction
H
The ability of humans to cope in high-altitude environments is enhanced through acclimatization, developmental adaptations, and evolutionary adaptations. The process of acclimatization involves transient adjustments that occur within an individual's lifetime (Houston et al., 1987; Bärtsch and Saltin, 2008; Martin et al., 2010); developmental adaptations involve irreversible but nonheritable adjustments in an individual arising from high-altitude exposures during periods of development (Frisancho, 2013); and evolutionary adaptations are irreversible and heritable adjustments that arise in populations following generations of exposure to high altitude (Moore, 2001; Beall 2007; Gilbert-Kawai, 2014). This review will focus on acclimatization and evolutionary adaptations to high-altitude environments.
There is marked variation in the physiological responses of humans to acute and chronic hypoxia exposures (e.g., cardiovascular, respiratory, and hematological responses) (Martin et al., 2010); however, the variation in response to hypoxia is perhaps most apparent in the differential susceptibility to acute and chronic altitude illnesses (MacInnis et al., 2010). Indeed, under identical hypoxic conditions, some high-altitude sojourners and residents will remain well while others will develop altitude illnesses, which can be potentially fatal.
Understanding the sources of variation in hypoxia acclimatization and adaptation is a major area of research for high-altitude biology (MacInnis et al., 2010; Simonson, 2015). In general, the concept that there is a genetic contribution to altitude illness susceptibility is supported by the apparent differences in susceptibility between populations, the notion that a history of altitude illness is indicative of subsequent risk, the heritability of hypoxia-related traits, and the association of specific genetic variants with susceptibility to altitude illness.
The primary focus of this review will be to present and evaluate the evidence for and against genetic predisposition to each of the major acute and chronic altitude illnesses. Since the previous edition of this review (MacInnis et al., 2010), substantial progress has been made in identifying genes with putative roles in altitude illness susceptibility through the use of genome-wide techniques. Accordingly, we will focus on data obtained from genome-wide association studies (GWAS) rather than candidate gene association studies (CGAS), which were the primary source of data for previous reviews (Rupert and Koehle, 2006; MacInnis et al., 2010). The reader is directed elsewhere for reviews of basic genetic terminology (Attia et al., 2009a; MacInnis et al., 2011), genetic association studies (Attia et al., 2009b, 2009c; Manolio, 2010), and the pathophysiology of altitude illness (Bartsch and Bailey, 2013; Bartsch and Swenson, 2013; Schoene and Swenson, 2013).
Testing for Genetic Predisposition to a Trait
A genetic predisposition (or inherited susceptibility) to a trait (e.g., altitude illness) is simply a greater chance of developing that trait because of the presence of one or more causal genetic variants. Evidence for and against a genetic predisposition to a particular altitude illness can be attained from various sources. First, for a trait to be considered genetic, the phenotypic variation in a population must be due to genetic variation (Hirschhorn and Daly, 2005; Visscher et al., 2008); however, without individual differences in susceptibility to altitude illnesses, it is difficult to discern the role of genetics, at least in that population. Second, susceptibility to altitude illness should be relatively stable within individuals, such that, under certain conditions, those who are susceptible always develop the illness and those who are not susceptible do not (Visscher et al., 2008); however, external forces (e.g., medicine, nutrition, and exercise) and longitudinal effects (e.g., changes in behavior and age) can modify the severity and/or presentation of many traits. Third, with all other factors being equal, the frequency of altitude illness susceptibility should differ in populations (e.g., biogeographic groups and families) with different frequencies of the causal genetic variant(s). For example, if susceptibility to altitude illness were genetic, it should aggregate in families, as family members are more genetically similar to each other than to nonrelatives.
Finally, specific genetic variants should be overrepresented in those who are susceptible to altitude illness compared with those who are not susceptible. Genetic association studies can involve several polymorphisms selected based on a priori hypotheses (i.e., CGAS) or thousands to millions of polymorphisms selected without specific a priori hypotheses (i.e., GWAS). While GWAS can potentially provide greater insights into traits for which the molecular mechanisms are undetermined, they typically require very large sample sizes, meaning that GWAS are relatively expensive and difficult to implement and interpret (Hirschhorn and Daly, 2005).
Understanding the genetic basis of a trait does not immediately (or necessarily) improve clinical outcomes. Even if those genes that conferred susceptibility to altitude illness were identified, the clinical utility of a genetic test would still have to be determined. In other words, it would be necessary to show that the information provided by the genetic test would improve health outcomes by directing individuals toward effective strategies to mitigate altitude illness.
Acute Altitude Illness
The term acute altitude illness generally encompasses three illnesses: acute mountain sickness (AMS), high-altitude pulmonary edema (HAPE), and high-altitude cerebral edema (HACE). Collectively, these illnesses affect millions of the high-altitude sojourners who travel above 2500 m each year, with consequences including interrupted travel, reduced productivity, and limited recreation. Without rapid medical intervention, HAPE and HACE can be lethal. Genetic differences have been postulated to explain individual variation in susceptibility to these conditions (MacInnis et al., 2010).
Acute mountain sickness
AMS: background
AMS is a transient condition developing at altitudes above 2500 m, characterized by nonspecific symptoms (headache, nausea, fatigue/weakness, dizziness/lightheadedness, insomnia) (Roach et al., 1993; Hackett and Roach, 2001). The exact physiological mechanisms that lead to the development of AMS are unknown (Bartsch and Bailey, 2013), but symptoms are hypothesized to arise due to cerebral perturbations resulting from hypoxia (Bailey et al., 2009; Bartsch and Bailey, 2013), whether normobaric or hypobaric (Roach et al., 1996; Richard et al., 2014). The incidence of AMS is highly variable, depending largely on the rate of ascent and the altitude attained (Maggiorini et al., 1990; Bloch et al., 2009). For example, a rapid ascent to moderate altitude causes most humans to develop AMS (e.g., a 3-hour drive from sea level to 4200 m) (Forster, 1985).
The presence/absence and severity of AMS are determined through self-report questionnaires (e.g., Lake Louise Score [LLS], Roach et al., 1993 and Environmental Symptom Questionnaire [ESQ-III], Sampson et al., 1983; Beidleman et al., 2007). While these questionnaires are relatively simple and rapid to use, the validity of each is difficult to establish without an objective gold standard for the assessment of AMS. Recent work from our research group (MacInnis et al., 2013b) and others (Hall et al., 2014) has demonstrated that the Lake Louise definition of AMS likely represents multiple clinical syndromes that occur on exposure to hypoxia, with impaired sleep quality appearing to be independent from other effects. These potential inconsistencies in the phenotype of AMS could introduce excessive variability into the assessment of the genetic contribution to this syndrome, complicating the interpretation of data.
AMS: individual susceptibility and repeatability
One of the traditional rationales for a genetic basis for AMS has been the notion that history is a useful predictor of susceptibility to AMS (Hackett and Roach, 2001; Imray et al., 2011; West, 2012). While history has been associated with a greater risk of developing AMS on occasion (Richalet et al., 2012), it was a poor predictor of AMS in a large meta-analysis (MacInnis et al., 2015a) (Fig. 1). Furthermore, across two blinded and identical normobaric hypoxia exposures (13% O2, ∼4000 m equivalent)—separated by at least 14 days—AMS severity was not repeatable (MacInnis et al., 2014). Other studies that reported AMS to be repeatable (Forster, 1984; Rexhaj et al., 2011) did not include sham trials, which is problematic for a condition measured with subjective self-reported symptoms. An individual's susceptibility to AMS might eventually stabilize with repeated exposures (e.g., mountaineers who frequently ascend to altitude); however, this hypothesis remains untested.
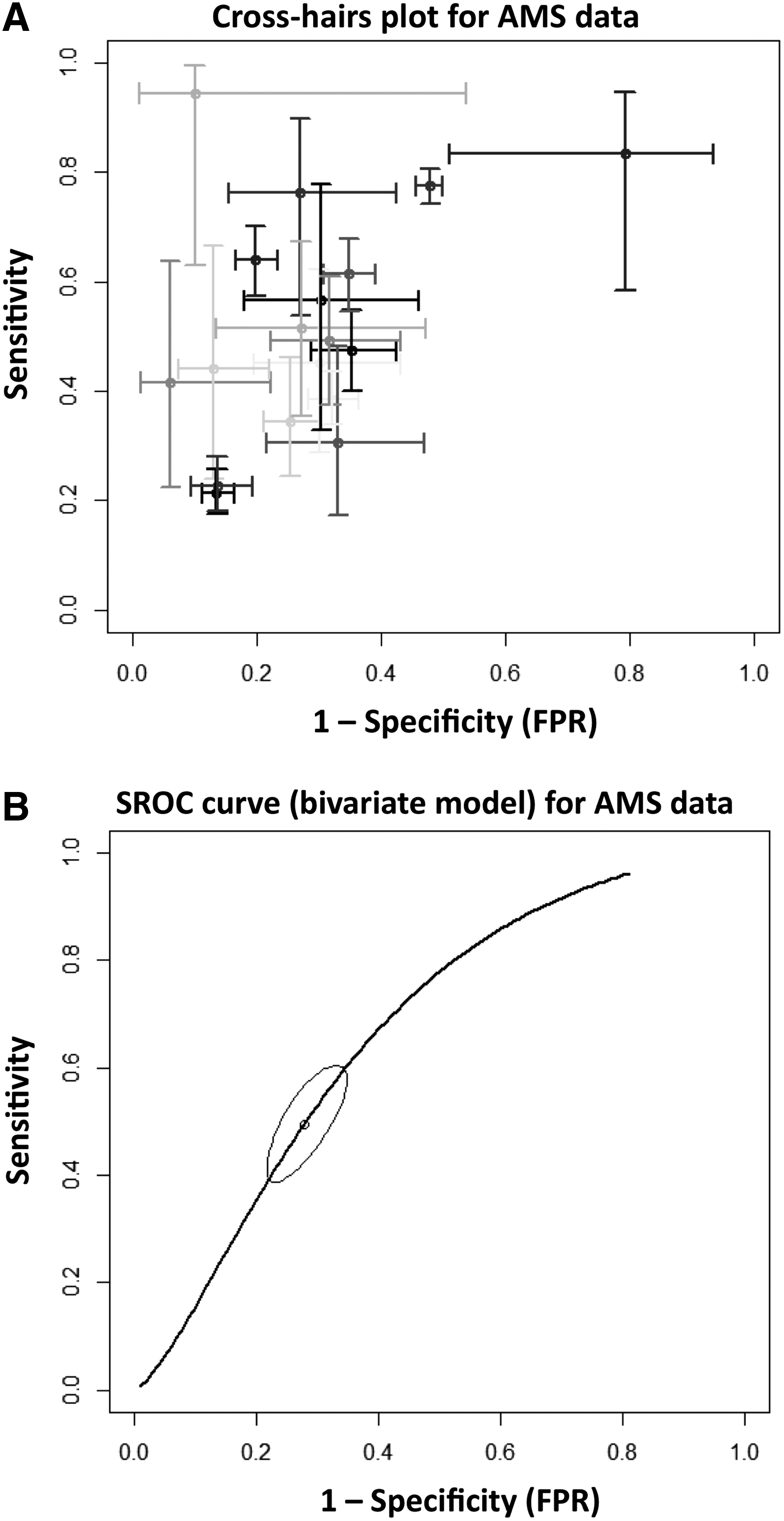
AMS: biogeographical and familial data
Despite its relatively high incidence, a dearth of biogeographical and family data is available for AMS susceptibility. Most biogeographical data compare Tibetans to other groups. For example, a large epidemiological study reported that, on exposure to ∼4500 m, Tibetans living at low altitudes had a much lower incidence of AMS than Han Chinese arriving from the same low altitudes (Wu et al., 2009). In a large prospective field study (MacInnis et al., 2013a), those with Tibetan ancestry (determined by surname) (Thornton et al., 2011) were at a lower risk of developing AMS than those of Indo-Caucasian ancestry on a rapid ascent to 4380 m (MacInnis and Koehle, Unpublished Data). Smaller anecdotal reports suggested that Tibetans and Sherpas are less likely to develop AMS than trekking groups of Han Chinese and Japanese ancestry (Wu et al., 2005). In all of these studies, it is difficult to account for the potentially confounding effects of environmental differences, such as health, diet, and cultural practices. To our knowledge, susceptibility to AMS has not been studied in Andeans, preventing biogeographical comparisons between Andeans and other populations.
Whether a family history of AMS increases one's risk of developing AMS is an unanswered question that should be addressed. In a large group of Nepalese pilgrims, the LLS of brothers was correlated, but insufficient familial data were available to make strong conclusions related to the predictive value of family history (MacInnis et al., 2013a). Twin studies suggest that AMS incidence is similar within pairs of twins, although only infants (Yaron et al., 2002) and children (Masschelein et al., 2014) have been studied. More research is needed to understand the influence of biogeographical group and family history on AMS susceptibility and whether either variable would be useful for predicting susceptibility.
AMS: genetic association studies
Numerous CGAS have tested associations between AMS susceptibility and specific genetic variants (reviewed by MacInnis et al., 2011) (Table 1); however, none of these studies has identified a genetic variant that has a strong association with AMS. For example, four separate studies (n = 103–284 subjects), which were performed on three different continents, reported that the angiotensin-converting enzyme (ACE) gene was not associated with AMS (Table 1).
Dehnert et al. (2002); 2Tsianos et al. (2005); 3Koehle et al. (2006); 4Kalson et al. (2008); 5Buroker et al. (2010); 6Kumar et al. (2004); 7Hotta, (2004); 8Rajput et al. (2006); 9Qi et al. (2007); 10Wang et al. (2013); 11Wu et al. (2015); 12Bhagi et al. (2015); 13Morrell et al. (1999); 14Aldashev et al. (2002); 15Aldashev (2007); 16Wang et al. (2007); 17Stobdan et al. (2010); 18Stobdan et al. (2011); 19Srivastava et al. (2012); 20Wang et al. (2010); 21Weiss et al. (2003); 22Ahsan et al. (2004); 23Zhang et al. (2014); 24Mejía et al. (2005); 25Mishra et al. (2012); 26Yang et al. (2013); 27Droma et al. (2003); 28Jiang et al. (2005); 29Mishra et al. (2011); 30Droma et al. (2008); 31Ding et al. (2011); 32Hanaoka et al. (1998); 33Li et al. (2004); 34Zhou et al. (2005); 35Qi et al. (2009); 36Aldashev (2007); 37Wang et al. (2009); 38Droma et al. (2002); 39Saxena et al. (2005); 40Hanaoka et al. (2003a); 41Ding et al. (2012); 42Hanaoka et al. (2003b).
AMS, acute mountain sickness; CMS, chronic mountain sickness; HAPE, high-altitude pulmonary edema; HAPH, high-altitude pulmonary hypertension.
More recently, we published the first GWAS on AMS susceptibility (MacInnis et al., 2015b). In that study, FAM149A (Table 2) was provisionally associated with AMS in a group of Nepalese pilgrims who rapidly ascended to 4380 m. This gene is highly expressed in the trigeminal and superior cervical ganglia (Su et al., 2004), tissues that are plausibly related to AMS pathophysiology (Bartsch and Bailey, 2013); however, in validation cohorts, the selected FAM149A polymorphism was not associated with AMS, suggesting that this association was potentially a false positive or that it has a relatively small effect size. No other genes were associated with AMS in that study.
Note that this study based the hypothesis on genomics data, but did not use a genomics approach.
Zhou et al. (2013); 2Cole et al. (2014); 3Mishra et al. (2015); 4Zhang et al. (2014); 5Mishra et al. (2012); 6Simonson et al. (2010); 7Aggarwal et al. (2010); 8Peng et al. (2011); 9Xu et al. (2011); 10Bigham et al. (2010); 11MacInnis et al. (2015b); 12Su et al. (2004); 13Wilkins et al. (2014); 14Cheng et al. (2007); 15Kobayashi et al. (2013); 16Loffek et al. (2011); 17Qi et al. (2003).
The hypoxia-inducible factor (HIF)-1α pathway has been implicated in AMS susceptibility. HIF-1α is an important transcription factor that regulates cellular homeostasis under conditions of hypoxic stress. Under normoxic conditions, prolyl hydroxylase 2 (PHD2), an oxygen sensor encoded by the EGLN1 gene, degrades HIF-1α. In several investigations into the genomic basis of high-altitude adaptation, particular EGLN1 variants have been overrepresented in Tibetans and Andeans (Aggarwal et al., 2010; Bigham et al., 2010; Simonson et al., 2010; Peng et al., 2011; Xu et al., 2011). Based on these studies, Zhang et al. (2014) examined the role of EGLN1 variants (Table 2) in AMS and reported that variants in the 5′-untranslated region of EGLN1 were associated with AMS in a relatively large sample of Han Chinese (190 cases, 190 controls); however, this association has not been replicated.
High-altitude cerebral edema
HACE: background
HACE is another cerebral form of altitude illness; however, unlike AMS, HACE is a rare encephalopathy. Some researchers suspect that HACE is a severe endpoint of AMS (Hackett and Roach, 2001) and AMS typically precedes HACE (Hackett and Roach, 2001, 2004); however, this is not always true, and HACE can occur without any symptoms of headache (Wu et al., 2006). A common pathophysiology for the two conditions has not been proven.
HACE usually occurs at least 2 days after ascent to an altitude above 3000 m (but usually above 4000 m) (Bartsch and Swenson, 2013). The incidence of HACE is relatively low, with estimates of 1% (Hackett et al., 1976) and 0.28% (Wu et al., 2007). Clinically, HACE is marked by ataxia of gait, severe lassitude, and changes in consciousness (Hackett and Roach, 2001, 2004). There is no questionnaire to diagnose HACE and other conditions can have similar presentations (e.g., central nervous system infection, hypoglycemia, hypothermia) (Hackett and Roach, 2001).
Magnetic resonance imaging studies of those with, or recovering from, HACE demonstrated vasogenic edema (Hackett et al., 1998) and microhemorrhages in the corpus callosum (Dickinson et al., 1983; Kallenberg et al., 2008; Schommer et al., 2013), indicating disruptions in the blood–brain barrier. Left untreated, HACE may progress to coma followed by death due to brain herniation within 24 hours (Bartsch and Swenson, 2013). To our knowledge, there have been no investigations into the potential genetic basis of HACE.
High-altitude pulmonary edema
HAPE: background
HAPE is a rare altitude illness. HAPE is a noncardiogenic form of pulmonary edema, secondary to exaggerated pulmonary artery pressure and hypoxic pulmonary vasoconstriction (Maggiorini et al., 2001; Swenson et al., 2002). These high vascular pressures are believed to cause the extravasation of fluid from pulmonary capillaries to alveolar spaces, which impairs diffusion in the lung, causing severe hypoxemia (Maggiorini et al., 2001; Swenson et al., 2002). Accordingly, HAPE is characterized by fatigue, breathlessness, coughing, frothy sputum, inspiratory crackles, cyanosis, tachypnea, and tachycardia (Hackett and Roach, 2001; Schoene and Swenson, 2013). The presence of patchy alveolar infiltrates on radiographs is an objective marker for HAPE.
The onset of HAPE is generally 2 days after arrival at a new altitude (usually above 3000 m) (Hackett and Roach, 2001; Schoene and Swenson, 2013). The risk of HAPE increases with ascent rate and the altitude attained. For example, in those with an unknown history of HAPE, the condition occurs in 0.2%, 2%, and 6% on 4-, 7-, and 1- or 2-day ascents to 4500 m and in 15% on a 1–2-day ascent to 5000 m (Bartsch and Swenson, 2013).
HAPE: individual susceptibility and repeatability
In some respects, HAPE is more amenable to research than AMS or HACE. First, there is clear individual susceptibility in HAPE: this illness is rare in those who have previously ascended to altitude without developing HAPE, but it is repeatable in those who have previously developed HAPE (i.e., risk of recurrence is ∼60% in those who ascend to 4500 m in 2 days) (Bartsch et al., 2002; Bartsch and Swenson, 2013). Second, HAPE has objective signs, which increase confidence in its diagnosis. Susceptible and nonsusceptible individuals can be accurately identified and studied prospectively with strong confidence in the quality of the phenotype data.
HAPE: biogeographical and familial data
Several instances of HAPE susceptibility within siblings and within parent–offspring pairs have been reported (Hultgren et al., 1961; Fred et al., 1962; Scoggin et al., 1977; Norboo et al., 2004; Lorenzo et al., 2009). The clinical utility of a family history of HAPE is unknown, but these cases of familial aggregation are supportive of a genetic etiology. To our knowledge, biogeographical comparisons are not available for HAPE.
HAPE: genetic association studies
Many candidate genes have been investigated as markers of HAPE susceptibility, but these studies have not identified any associations with clinical utility (Table 1); however, two GWAS have recently identified several candidate genes for HAPE susceptibility that merit further investigation.
The first of these studies, from Kobayashi et al. (2013), reported an association between the tissue inhibitor of metalloproteinase 3 gene (TIMP3; Table 2) and HAPE in a Japanese population. TIMP3 regulates the degradation of the extracellular matrix of lung tissue, and the interaction of TIMP proteins and matrix metalloproteinases has been associated with various lung pathologies related to decreased structural integrity (e.g., edema, emphysema, fibrosis) (Loffek et al., 2011). Although this finding is intriguing, it has not yet been replicated and was not identified in another similar study of HAPE.
The second GWAS for HAPE susceptibility compared the frequency of genetic variants among HAPE-susceptible subjects, HAPE-resistant subjects, and native highlanders. From the initial analysis, putative associations were found between HAPE susceptibility and variants of apelin (APLN), apelin receptor (APLNR), and nitric oxide synthase 3 (NOS3) (Mishra et al., 2015) (Table 2). Apelin induces vasodilation by increasing nitric oxide production via a NOS3 pathway, and in hypoxic conditions, HIF increases apelin expression, which augments vasodilation. In follow-up assays, apelin-13 and nitrite concentrations were lower in the blood of HAPE-susceptible subjects relative to HAPE-resistant subjects, which corresponded with (1) the lower gene expression of APLN, APLNR, and NOS3 in HAPE-susceptible subjects, (2) the higher methylation of APLN CpG islands in HAPE-susceptible subjects, and (3) the results of an APLN promoter assay.
As with AMS, the association of EGLN1 with high-altitude adaptation has prompted investigations into its potential role in HAPE susceptibility (Table 2). First, Aggarwal et al. (2010) identified two variants of EGLN1, which were associated with its expression, that were
While these genetic studies of HAPE are intriguing, further work is needed to confirm their validity (i.e., these associations have not been independently replicated). Furthermore, whether these associations can be generalized to other populations and whether they have clinical utility have not been established.
Chronic Altitude Illness
Chronic altitude illnesses affect residents of high-altitude environments, whether they are temporary residents, long-term residents, or multigeneration high-altitude natives (León-Velarde and Villafuerte, 2011; Leon Velarde et al., 2014). The major chronic altitude illnesses are chronic mountain sickness (CMS) and high-altitude pulmonary hypertension (HAPH). While much of the focus of high-altitude biology has been on the successful adaptation of high-altitude natives to hypoxia (e.g., Tibetans, Andeans, and Ethiopians), efforts have also been made to explain the individual variation in susceptibility to chronic altitude illness within these biogeographical groups.
Chronic mountain sickness
CMS: background
CMS affects high-altitude natives and long-time residents of high altitude (León-Velarde et al., 2005). CMS generally develops after years of exposure to high altitude (≥2500 m) and it resolves with descent to lower altitudes (León-Velarde et al., 2005). Excessive erythrocytosis is the primary sign of CMS, but severe hypoxemia and pulmonary hypertension may also be present (León-Velarde et al., 2005). The pulmonary hypertension associated with CMS can cause right-heart failure and congestive heart failure (Leon Velarde et al., 2014).
The presence and severity of CMS are ascertained through a questionnaire. The signs and symptoms of interest are breathlessness/palpitations, sleep disturbance, cyanosis, dilatation of veins, paresthesia, headache, tinnitus, and hemoglobin concentration (León-Velarde et al., 2005). To confidently diagnose CMS, diseases with similar presentations (e.g., chronic pulmonary diseases) must be excluded (León-Velarde et al., 2005).
CMS: individual susceptibility and repeatability
Despite being exposed to the same conditions, only some highlanders develop CMS. The prevalence ranges from 0% to 5% among native highlanders in Asia, Africa, and South America. To treat CMS, subjects typically require ongoing venesection. Symptoms typically resolve with descent to low altitude, but they recur on reascent to high altitude (León-Velarde et al., 2005). These points demonstrate that there is individual susceptibility to CMS and that susceptibility to CMS is relatively stable.
CMS: biogeographical and familial data
The prevalence of CMS varies geographically. Andeans are more likely to have CMS than Tibetans (∼5% vs. <1%, respectively), despite occupying similar altitudes (Moore, 2001; Leon Velarde et al., 2014). The lower incidence in Tibetans than Andeans might be a consequence of Tibetans' generally higher alveolar ventilation and hypoxic ventilatory response and lower hemoglobin concentration (Beall et al., 1997, 1998; Moore, 2000; Beall, 2007). Having spent generations at high altitude seems to decrease the likelihood of CMS, as the prevalence of CMS in Tibetan highlanders is much lower than that of Han Chinese immigrants who occupy the same altitudes (Moore, 2001). Familial data related to CMS are surprisingly rare; however, an early study reported familial aggregation of CMS in a South American population (Reátegui, 1969). CMS has not been reported in Ethiopian highlanders (Leon Velarde et al., 2014).
CMS: genetic association studies
Several CGAS have been performed on Andeans with and without CMS, but these studies have not identified a strong candidate gene to explain the differential susceptibility to CMS among Andeans (Table 1). A recent study that sequenced whole genomes of 10 individuals with CMS and 10 individuals without CMS identified several genomic regions of interest (Zhou et al., 2013). Genes from two of these regions (SENP1 and ANP32D; Table 2) had greater expression in CMS-derived cultured fibroblast cells relative to non-CMS-derived cells and decreasing the expression of these genes in flies increased their survival when exposed to hypoxia. One of the identified genes, SENP1, regulates erythropoiesis (Cheng et al., 2007; Yu et al., 2010), suggesting that its upregulation could contribute to the erythrocytosis seen in CMS. The putative role of ANP32D in the pathophysiology of CMS is unknown.
These results were partially replicated: SENP1 was, but ANP32D was not, associated with CMS in two cohorts of Andean highlanders from Cerro de Pasco, Peru (Cole et al., 2014). The sample size of the replication study (84 cases; 91 controls) was much larger than the original study, and the CMS patients from the two studies had similar degrees of erythrocytosis. Given that the two genes were in linkage disequilibrium and the potential role of ANP32D is unclear, Cole et al. suggested that SENP1 could be driving the association.
Andeans with CMS are not a homogeneous group. Recent work from Villafuerte et al. (2014) demonstrated erythropoietin (EPO) concentrations to be elevated in some, but not all, Andeans with CMS. The authors suggested these phenotypes could represent genetically distinct subtypes of CMS; however, several genes related to the EPO pathway were not associated with CMS in previous CGAS (Table 1). More work is needed to understand whether genetic differences in genes related to the EPO pathway contribute to variation in CMS susceptibility.
High-altitude pulmonary hypertension
HAPH: background
HAPH is a chronic form of altitude illness that can develop in high-altitude natives and lifelong, high-altitude residents following prolonged stays above 2500 m (León-Velarde et al., 2005). HAPH is known by many names, including CMS of the vascular type, high-altitude heart disease, and subacute mountain sickness (León-Velarde et al., 2005). HAPH is characterized by excessive pulmonary artery pressure as well as elevated pulmonary vasoconstriction and pulmonary artery pressure (León-Velarde et al., 2005). Pulmonary vasculature remodeling, right ventricular hypertrophy, and congestive right-heart failure may occur in patients with HAPH (León-Velarde and Villafuerte, 2011). HAPH can occur independent of, or in conjunction with, CMS (Penaloza and Sime, 1971). To diagnose HAPH, other forms of pulmonary hypertension as well as chronic obstructive pulmonary diseases, interstitial diseases, and cardiovascular diseases must be excluded (León-Velarde et al., 2005).
HAPH: individual susceptibility and repeatability
Similar to CMS, only some highlanders develop HAPH. In a group of Kyrgyz highlanders, 23% of males and 6% of females had ECG signs of cor pulmonale (Aldashev et al., 2002), which is likely an overestimation of the incidence of HAPH, as only those with exertional dyspnea were screened. While pulmonary artery pressure decreases on descent to lower altitude, it rises to pathological values on return to high altitude in those with HAPH (Wilkins et al., 2014), suggesting HAPH is relatively stable.
HAPH: biogeographical and familial data
Tibetans generally have less pulmonary hypertension than Andeans (Groves et al., 1993), but to our knowledge, direct comparisons of HAPH incidences across biogeographical groups have not been made. For example, Tibetans living at 3658 m had a mean pulmonary artery pressure of 15 mmHg, whereas the corresponding values from Andeans living at ∼3700 m ranged from 20 to 23 mmHg (reviewed in Groves, et al., 1993). High-altitude heart disease (i.e., HAPH) was reported in multiple family members from three separate families living at high-altitude in China (Ge and Helun, 2001). The relatively stable nature of HAPH, like CMS, would facilitate more familial and biogeographical studies.
HAPH: genetic association studies
Most of the HAPH genetics studies to date have been CGAS, and relatively few genes have been investigated (Table 1). Although some variants have been statistically associated with HAPH, their clinical utility is unclear (MacInnis et al., 2010). A recent exome sequencing study of Kyrgyz highlanders identified GUCY1A3 as a candidate gene for HAPH susceptibility (Table 2) (Wilkins et al., 2014). That study revealed a rare missense mutation of the GUCY1A3 gene in three individuals with normal pulmonary artery pressure (and in 0/28 of those with elevated pulmonary artery pressure). This variant altered the activity of the soluble guanylyl cyclase enzyme in functional assays (Wilkins et al., 2014), suggesting that the missense mutation enhances sensitivity to nitric oxide, a vasodilating compound, to potentially reduce pulmonary hypertension. This association cannot fully explain the differential susceptibility to HAPH in the entire sample; however, this result is an example of the potential contribution of relatively rare variants to the variability in traits (Manolio et al., 2009).
Advancing Our Understanding of Altitude Illness: Summaries and Challenges
The purpose of this review was to (1) summarize and weigh the evidence for the genetic basis of altitude illness susceptibility and (2) highlight the recent progress in our understanding of the genetic contributors to altitude illnesses. Since our most recent review in this area (MacInnis et al., 2010), progress in understanding the extent to which genetic variability influences altitude illness susceptibility has been limited. In general, more data are needed to understand the repeatability/stability of each altitude illness and the extent to which altitude illness susceptibility aggregates in families and differs across biogeographical groups. Whereas CGAS studies were predominant in the past, genome-wide techniques now dominate the research. This shift to a genomics-based approach has proved advantageous for HAPE, CMS, and HAPH, but work is still needed to validate provisionally associated genes and determine their clinical utility.
Relatively little new information relating to the genetic basis of AMS has been published in the past 6 years. In our opinion, the current paucity of evidence for a genetic predisposition for AMS might have more to do with the quality of phenotype data than the influence of genetics on hypoxia tolerance per se. Some clarity is necessary in terms of the diagnosis and classification of AMS before investigators can more definitively quantify its heritability and better investigate its genetic determinants.
Our understanding of genetic susceptibility to AMS is hindered by the unclear pathophysiology of AMS (Hall et al., 2014; Luks, 2014), the low repeatability of AMS (MacInnis et al., 2014, 2015a), and the subjective nature of the current AMS criteria (Roach et al., 1993). As the pathophysiology is unclear and symptoms could arise from multiple pathways, researchers should consider examining high-altitude headache and aspects of oxygen transport/delivery that might contribute to hypoxia acclimatization (MacLeod et al., 2013).
As with AMS, there has been minimal progress in our understanding of the genetic basis of HACE. To our knowledge, there are no published reports of individuals suffering repeated episodes of HACE, biogeographical or familial aggregation of HACE, or genetic association studies of HACE. Compared to AMS, the HACE phenotype is better defined, with imaging of the brain permitting objective diagnoses; however, HACE research is restricted by its rare and severe nature. Obtaining sufficient sample sizes to perform genetic association studies would likely require the establishment of HACE databases that track cases from large geographical areas or international collaborations that share data. Investigations of HACE might be well informed by animal models (Huang et al., 2015).
Among the acute altitude illnesses, genetic predisposition to HAPE susceptibility has the strongest support. Opportunistic field studies are difficult to implement with HAPE because of its relatively low incidence (and relatively longer time to onset than AMS); however, because of its strong repeatability and the high confidence in diagnosis, the establishment of a HAPE registry could prove a useful tool to study HAPE susceptibility from a genetic perspective. More family studies, including prospective data on the clinical utility of a familial history of HAPE, would likely be very informative (and useful, from an applied perspective).
Five genes have been identified that appear to affect HAPE susceptibility, either through structural changes in the lung, through the regulation of the HIF pathway, or through apelin and nitric oxide signaling pathways. Studies have not replicated these associations, so more work is needed before this information can better inform our understanding of HAPE pathophysiology and susceptibility.
The genetic work from Zhou et al. (2013) has identified intriguing candidate genes for CMS, and in our opinion, genetic susceptibility to CMS has the best support of all the altitude illnesses: SENP1 was identified through a genome-wide analysis, SENP1 has a putative role in the pathophysiology of CMS, and the association between SENP1 and CMS was independently replicated. Further attempts at replicating and understanding this association are warranted. While the differential susceptibility between Han Chinese, Tibetan, and Andean populations is suggestive of a genetic basis to CMS, it is not conclusive. Familial data within each highland population could help to determine the extent to which CMS is an inherited condition, and the chronic and stable nature of CMS would facilitate these types of studies.
The first genome-wide study of HAPH has identified a candidate gene with a plausible role in HAPH; however, unlike CMS, this association has not been replicated. More studies—biogeographical, family, and genetic—are needed to determine whether HAPH susceptibility has a strong genetic component and to confirm the association between HAPH and GUCY1A3. Because HAPH is a relatively rare condition that occurs in remote populations, large studies are difficult. Attempting to identify the genes influencing variation in the pulmonary artery pressure response to hypoxic gas in lowlanders could be beneficial, as it could alleviate these issues.
In summary, over the past 6 years, there has been some progress in understanding the genetic underpinnings of altitude illness, but the search for genes that influence altitude illness susceptibility is proving more difficult than previously anticipated (MacInnis et al., 2010). The main challenges appear to be inconsistent stimuli (e.g., ascent rate, weather, and altitude achieved), the rarity of the conditions, the remoteness of the affected populations (particularly high-altitude natives), and inconsistencies and uncertainties in the phenotypes. Progress has been greatest for those conditions that are repeatable and stable with objective signs (i.e., HAPE, CMS, HAPH).
Understanding the influence of genetic differences on altitude illness susceptibility may ultimately help with the treatment and management of these potentially life-threatening illnesses; however, an improved understanding of high-altitude physiology and adaptation is the more likely and more immediate outcome of continued research in this area.
Footnotes
Author Disclosure Statement
No competing financial interests exist.