
Abstract
Abstract
Fan, Jui-Lin, and Bengt Kayser. Fatigue and exhaustion in hypoxia: the role of cerebral oxygenation. High Alt Med Biol. 17:72–84, 2016.—It is well established that ascent to high altitude is detrimental to one's aerobic capacity and exercise performance. However, despite more than a century of research on the effects of hypoxia on exercise performance, the underlying mechanisms remain incompletely understood. While the cessation of exercise, or the reduction of its intensity, at exhaustion, implies reduced motor recruitment by the central nervous system, the mechanisms leading up to this muscular derecruitment remain elusive. During exercise in normoxia and moderate hypoxia (∼1500–2500 m), peripheral fatigue and activation of muscle afferents probably play a major role in limiting exercise performance. Meanwhile, studies suggested that cerebral tissue deoxygenation may play a pivotal role in impairing aerobic capacity during exercise in more severe hypoxic conditions (∼4500–6000 m). However, recent studies using end-tidal CO2 clamping, to improve cerebral tissue oxygenation during exercise in hypoxia, failed to demonstrate an improvement in exercise performance. In light of these recent findings, which seem to contradict the hypothetical role of cerebral tissue deoxygenation as a performance limiting factor at high altitude, this short review aims to provide a critical reappraisal of the extant literature and ends exploring some potential avenues for further research in this field.
Introduction
H
It seems apparent that the cessation, or the reduction in intensity, of a fatiguing exercise task is mediated by the brain (Kayser, 2003). Various factors, such as sensory feedback from the working limbs (Amann et al., 2008, 2011) or the respiratory muscles (Amann et al., 2007a), circulating metabolites (Karlsson et al., 1975; Hogan and Welch, 1984), arterial O2 content (CaO2) and cardiac output (Amann, 2006; Amann et al., 2006), the body's glycogen stores (Bergstrom et al., 1967; Hermansen et al., 1967), and thermal stress (Nybo and Nielsen, 2001; Nybo et al., 2002), can contribute toward the development of fatigue, but the cessation of exercise is ultimately initiated by the brain (Kayser, 2003). From this perspective, the endpoint of increasing fatigue (i.e., exhaustion) is of central nervous system (CNS) origin (Kayser, 2003; Secher et al., 2008; Noakes, 2012).
Aerobic exercise capacity is highly sensitive to changes in SaO2 (Amann and Kayser, 2009), and even in conditions of normoxia, exercise-induced arterial hypoxemia impairs exercise capacity [see Dempsey and Wagner (1999) for review]. This detrimental effect of arterial hypoxemia on aerobic capacity is exacerbated during exercise in hypoxia (Kayser et al., 1994; Amann and Calbet, 2008). As Amann and Kayser (2009) noted, this impairment of aerobic capacity, during both acute and chronic exposure to high altitude, has been well documented in both moderately and highly trained individuals (Dill et al., 1966, 1967; Klausen et al., 1966; Maher et al., 1974; Calbet et al., 2003; Lundby et al., 2004, 2006). Furthermore, it appears that people with higher aerobic fitness at sea level are more adversely affected in hypoxia (Martin and O'Kroy, 1993; Gore et al., 1996; Ferretti et al., 1997; Mollard et al., 2007; Calbet and Lundby, 2009), due to greater reductions in SaO2, CaO2, and therefore mass O2 transport (Ekblom et al., 1975; Chapman et al., 1999; Calbet and Lundby, 2009). This reduction in mass O2 transport ultimately leads to an early onset of fatigue and exhaustion compared to equivalent exercise at lower altitudes (Amann and Kayser, 2009).
Fatigue can be defined as an exercise-induced loss of capacity of muscle force and/or velocity production, which is reversible by rest (Bigland-Ritchie and Woods, 1984; Gandevia, 2001). Traditionally, it was thought that dynamic exercise with large muscle groups such as cycling is limited by the development of muscular anaerobiosis, resulting in an inability of the exercising muscles to produce further contractions. This notion was first proposed by Nobel Laureate A.V. Hill in 1923. Despite seminal early work by Angelo Mosso who proposed a role for the CNS in neuromuscular fatigue [reviewed in Gandevia (2001)], for a long time the notion that fatigue during dynamic exercise originates from the skeletal muscles dominated the field of exercise science [see Noakes (2011) for a historical review]. The prevailing model now is that neuromuscular fatigue can be explained by changes all along the chain from central command down to the myofibrils. A distinction is made between peripheral fatigue (i.e., in the muscle) and central fatigue (i.e., in the CNS down to the motor end plate). Today there is ample evidence that supports a role for the CNS also for modulating dynamic exercise performance (Gandevia, 2001; Noakes, 2011). In this article, we provide a short review of the current literature concerning the roles for peripheral and central fatigue during dynamic exercise with large muscle groups in hypoxia. Furthermore, we critique recent strategies to manipulate central fatigue during exercise in hypoxia and propose future avenues to unravel its elusive origin.
Peripheral Fatigue
The major causes of fatigue development during muscle contraction have been attributed to muscle contractile apparatus failure, impairment of excitation–contraction coupling mechanisms associated with excess accumulation of metabolic by-products, and impaired sarcoplasmic reticulum Ca2+ handling (Merton, 1954; Dawson et al., 1978; Fitch and McComas, 1985; Vollestad et al., 1988; MacIntosh et al., 2012; Ortenblad et al., 2013). Accordingly, it was hypothesized that greater metabolic disturbance associated with reduced oxygen availability, at the level of the muscle fiber, could be responsible for the reduced exercise performance observed in severe hypoxia (Bylund-Fellenius et al., 1981; Idstrom et al., 1986). However, evidence from Operation Everest II, a hypobaric chamber simulation of a Mount Everest ascent, questioned this contention. At extreme simulated altitude metabolic enzyme activity, muscle oxidative capacity, and energy stores in the vastus lateralis at the end of exhaustive cycling exercise were all largely preserved (Green et al., 1989). Furthermore, in the same subjects, electrical stimulation was able to elicit full ankle dorsiflexion under extreme hypoxic conditions, suggesting that muscle contractility per se was preserved (Garner et al., 1990). Since biochemical, electromyographic, and mechanical signs of muscle fatigue at exhaustion were lower compared to normoxia, Verges et al. (2012) summarized that the main cause of impaired whole body exercise performance in severe hypoxia is unlikely due to muscle metabolic fatigue. Instead, as proposed by Bigland-Ritchie and Vollestad (1988) and then empirically suggested by Kayser et al. (1994), the CNS might play a primary role in limiting large muscle group performance under severe hypoxic conditions. Supporting this contention, Amann et al. (2007b) found smaller reductions in potentiated quadriceps twitch force—index of peripheral fatigue—at the end of constant-load cycling to exhaustion (∼80% normoxic power output) in severe hypoxia (fraction of inspired O2 [FIO2]: 0.10), compared to normoxia and moderate hypoxia (FIO2: 0.15) (Fig. 1). From this, they concluded that peripheral muscle fatigue is a main determinant of central motor drive and exercise performance in normoxia and moderate hypoxia (Amann et al., 2006; Romer et al., 2007), while a hypoxia-sensitive CNS component might play a greater role during high-intensity exhaustive exercise in more severe hypoxia.

Peripheral quadriceps fatigue (Qtw,pot, potentiated twitch force elicited by magnetic nerve stimulation) assessed 2 minutes following cycling exercises of various duration while breathing air with different values of FIO2. *Different from FIO2: 0.15 and 0.30 (p < 0.05); †different from FIO2: 0.10, 0.15, and 0.30 (p < 0.05). From Amann et al. (2007b). FIO2, fraction of inspired O2.
The volume of activated muscle appears to influence the degree of peripheral fatigue. Under normoxic conditions, studies have found greater reductions in potentiated quadriceps twitch force during single-leg knee extension to exhaustion compared to two-legged knee extension or cycling exercise (Rossman et al., 2012, 2014). From this, the authors concluded that the fatigue is predominately peripheral in origin during exercise involving smaller muscle mass. At 5050 m, Kayser et al. (1994) found similar performance and level of peripheral fatigue (from EMG) during dynamic arm exercise compared to sea level, while cycling exercise until exhaustion was accompanied by peripheral fatigue at sea level only, suggesting a minor role for peripheral metabolic changes for the cessation of exhaustive large muscle group exercise at high altitude.
Accelerated peripheral fatigue development in hypoxia
Following exhaustive constant-load cycling (∼50% normoxic power output) in acute hypoxia (FIO2: 0.105), Amann et al. (2013) found that both potentiated quadriceps twitch force and maximal voluntary contraction force were reduced compared to pre-exercise values (Fig. 2A). In contrast, they found no sign of peripheral fatigue following an identical bout of exercise in normoxia. Since inspiratory muscle work was higher and CaO2 was lower during hypoxic exercise compared to normoxia (Fig. 2B), the authors attributed the development of peripheral fatigue in hypoxia to reduced muscle oxygen delivery (DO2) and the increased respiratory work associated with hypoxic exercise (Fig. 3). Increased respiratory work, either by voluntarily increasing the tidal integral of transdiaphragmatic pressure or by hypoxia-induced hyperventilation, exacerbates diaphragm fatigue during exercise (Babcock et al., 1995; Vogiatzis et al., 2008). Diaphragm fatigue (and other respiratory muscle fatigue) is accompanied by metabolite accumulation and activation of unmyelinated group IV phrenic afferents (respiratory metaboreceptors) (Dempsey et al., 2006); these in turn increase sympathetic nerve activity (St Croix et al., 2000) and activate the respiratory muscle metaboreflex (Hussain et al., 1991; Sheel et al., 2001, 2002). This supraspinal reflex elicits constriction of the active locomotor muscle vasculature (Dempsey et al., 2006), redistributing cardiac output from the locomotor muscles to the respiratory muscles (the “blood steal” effect) (Harms et al., 1997, 1998). As a result, increased respiratory work during exercise causes earlier respiratory muscle fatigue onset and lowers locomotor muscle blood flow, thereby increasing the extent of locomotor muscle fatigue and perceptions of leg discomfort (Romer et al., 2006; Taylor and Romer, 2008). These effects of increased respiratory work on muscle DO2, muscle fatigue, and perceptions of leg discomfort are exacerbated by hypoxia (Amann et al., 2007a; Verges et al., 2010). Collectively, these findings demonstrate that muscle DO2 can be impaired through increased respiratory work and the associated metaboreflex-mediated reduction in locomotor limb blood flow during large muscle group exercise in hypoxia (Fig. 3).


Schematic representation of the effect of hypoxia on central motor drive. Respiratory system: Hypoxia lowers PaO2, CaO2, and respiratory muscle DO2 and stimulates ventilatory drive (Amann et al., 2007a). The combination of increased ventilation and reduced respiratory muscle DO2 accelerates the development of respiratory muscle fatigue, resulting in activation of respiratory muscle metaboreceptors (Dempsey et al., 2006). Locomotor muscles: Increases in both carotid chemoreceptor activity and respiratory metaboreceptor activity elicit vasoconstriction of the locomotor vasculature, thereby lowering limb blood flow (Dempsey et al., 2006). The reduced limb blood flow, combined with reduced CaO2 in hypoxia, lowers locomotor muscle DO2, augments the accumulation of metabolites, and accelerates the development of peripheral fatigue (Amann and Calbet, 2008). Consequently, the increase in peripheral fatigue modulates central motor drive through group III/IV muscle afferents (Amann et al., 2006). Brain: Increases in central motor drive associated with exercise elevate regional neuronal activity, and therefore, metabolic requirements (Raichle et al., 1976). These, in turn, elicit local vasodilation through neurovascular coupling, thereby increasing local blood flow and DO2 (Jorgensen et al., 1992a, 1992b). The increased ventilatory drive associated with hypoxia exacerbates hypocapnia and associated cerebral vasoconstriction. This cerebral vasoconstriction is partly alleviated by hypoxemia-mediated vasodilation and cortical activation-mediated vasodilation associated with exercise. The resulting reduction in cerebral blood flow, coupled with reduced CaO2, lowers cerebral DO2 and tissue oxygenation (Subudhi et al., 2007b). The subsequent reduction in cerebral tissue oxygenation, combined with increased group III/IV phrenic and muscle afferents' discharge, exacerbates the development of central fatigue and lowers central motor drive during exercise in hypoxia (Amann and Dempsey, 2008; Amann et al., 2013). CaO2, arterial O2 content; DO2, oxygen delivery.
Role of group III/IV afferent feedback on fatigue
It has been postulated that metabolic disturbances in the working muscle and subsequent development of central fatigue are modulated by group III/IV muscle afferent feedback, through inhibition of central motor drive (Fig. 3) (Amann and Dempsey, 2008; Amann, 2012; Rossman et al., 2012; Kennedy et al., 2015). This contention is supported by the observation that group III/IV muscle afferent blockade exacerbates peripheral fatigue development and impairs exercise performance (Amann et al., 2011). However, recent findings during whole-body exercise suggest that the role of group III/IV afferent feedback on exercise capacity is even more complex (Morales-Alamo et al., 2015; Torres-Peralta et al., 2015).
Using bilateral thigh occlusion (10 and 60 seconds) immediately following incremental exercise to exhaustion in normoxia and hypoxia (FIO2: ∼0.10), Torres-Peralta et al. (2015) reported greater muscle activation during subsequent isokinetic sprint compared to that observed during the last 10 seconds of the incremental cycling in both conditions. Since metabolite accumulation at exhaustion and during ischemia augments III/IV muscle afferent discharge (Darques et al., 1998; Light et al., 2008), these findings led the authors to conclude that group III/IV afferent feedback does not play a significant role in limiting sprint exercise performance under both normoxic and hypoxic conditions.
Intriguingly, they found better sprint performance, as indicated by peak and mean power output, following 60 seconds of thigh ischemia compared to 10 seconds of ischemia. As a more prolonged limb ischemia (60 seconds vs. 10 seconds) should lead to a reduced sprint performance due to greater metabolite accumulation, these findings provide further evidence against the limiting role of muscle metaboreflex (and therefore muscle III/IV afferents) in exercise performance. Instead, the authors postulated that the improved sprint performance following prolonged limb ischemia could be due to greater recovery of central mechanisms of fatigue. Alternatively, some partial peripheral recovery may have taken place despite the ischemia, possibly due to a shift of resources (oxygen, fuel) from less activated tissue to fatigued muscles, and metabolites (H+, lactate) in the opposite direction.
Thus, while moderate hypoxia appears to exacerbate the development of peripheral fatigue, reports of reduced peripheral fatigue following exercise to exhaustion in severe hypoxia, despite greater performance impairment, point to a different mechanism of exercise cessation. Findings from operation Everest II provided early clues that the inputs responsible for an early termination of exercise in severe hypoxia originated from above the level of the muscle (Bigland-Ritchie and Vollestad, 1988). Subsequently, work by Kayser et al. (1994) led many to speculate that the CNS must play a crucial role in limiting performance during large group exercise in severe hypoxia, possibly to maintain sufficient DO2 to the brain.
Central Fatigue
It has been proposed that in severe hypoxic conditions, the lack of oxygen for the CNS would lead to a reduced central motor drive, thus limiting exercise performance (Kayser et al., 1994; Amann et al., 2007b; Amann and Calbet, 2008; Amann and Kayser, 2009). Reduced cerebral DO2 and subsequent cerebral tissue deoxygenation could be a major limiting factor of exercise performance in hypoxia (Kjaer et al., 1999; Amann et al., 2006, 2007b; Subudhi et al., 2007b; Rasmussen et al., 2010b; Vogiatzis et al., 2011). An indication that such may be the case was reported by Kayser et al. (1994), who were the first to demonstrate that exhaustive exercise at 5050 m could be prolonged with a rapid increase in FIO2 (FIO2: 1.0) at the point of maximal exertion, in acclimatized subjects during constant-load cycling to exhaustion. This observation has since been repeated during both constant-load exercise to exhaustion (Amann et al., 2007b) and incremental cycling (Subudhi et al., 2007b; Koglin and Kayser, 2013). These later studies further demonstrated that rapid switching to hyperoxia (FIO2: 0.30–0.60) improved performance during exercise in acute and chronic hypoxia, while no improvement could be found by increasing FIO2 at exhaustion in normoxia (Fig. 4). Since the effect of the FIO2 increase was too quick to reverse the metabolic factors associated with peripheral fatigue, Kayser et al. (1994) and others (Amann et al., 2007b; Subudhi et al., 2007b; Koglin and Kayser, 2013) concluded that the improvement in exercise performance was due to cerebral tissue reoxygenation. However, as noted by Subudhi et al. (2011), these conclusions based on rapid O2 switching studies are limited by the fact that the improvement of arterial oxygenation is systemic, and thus, not localized only to the brain.
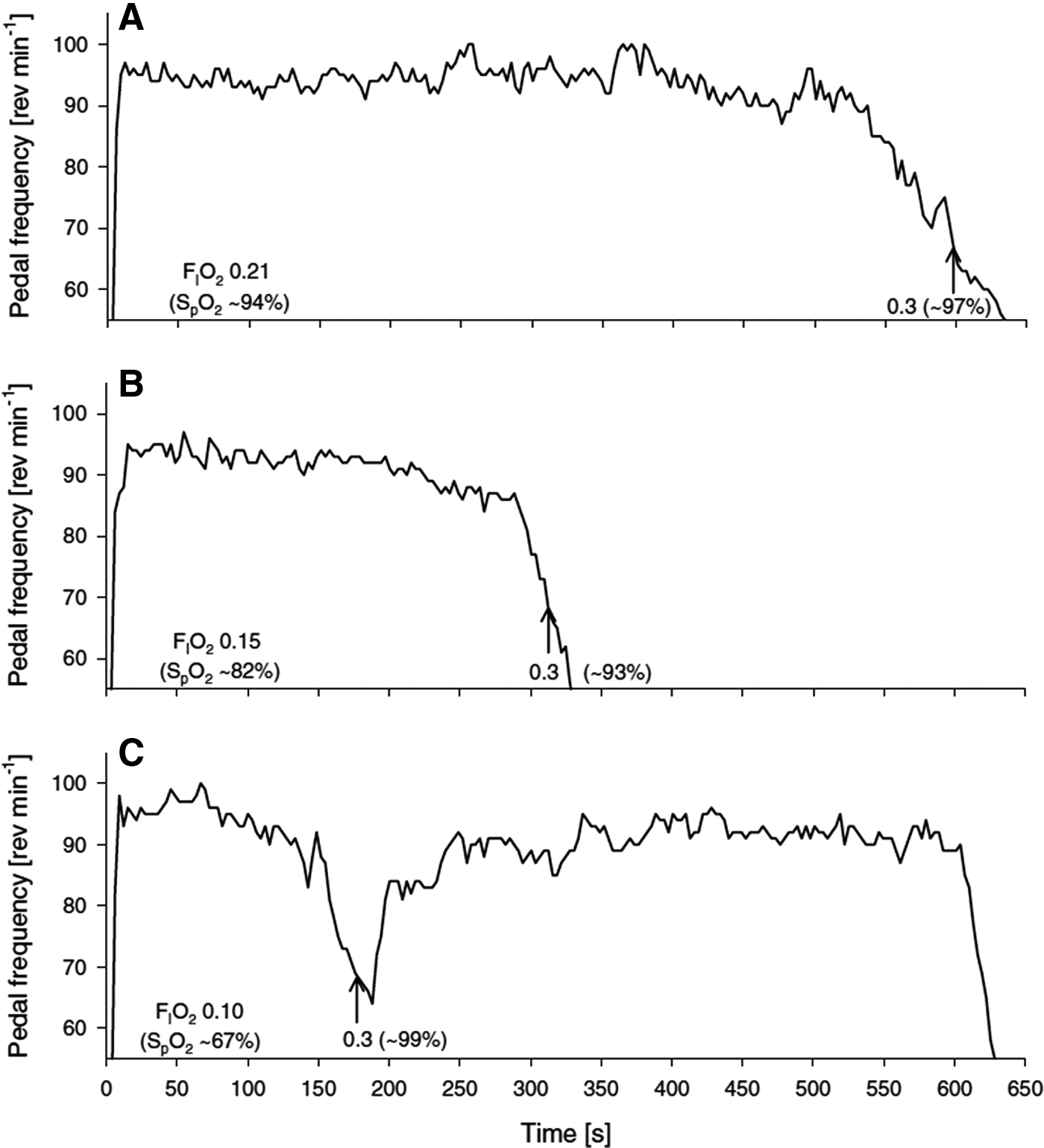
Representative example showing the effects of a rapid increase in FIO2 at exhaustion on pedal frequency and endurance time to exhaustion. The arrow indicates the point at which the inspirate was switched from normoxia
Limiting role of cerebral oxygenation
During high-intensity exercise, hyperventilation-induced hypocapnia leads to cerebral vasoconstriction (Herholz et al., 1987; Thomas et al., 1989; Jorgensen et al., 1992b; Madsen et al., 1993; Subudhi et al., 2007a). Under hypoxic conditions, this cerebral vasoconstriction counteracts the hypoxia-induced vasodilation, thereby lowering cerebral blood flow, cerebral DO2, and cerebral oxygenation (Fig. 3). An enhanced ventilatory drive associated with hypoxia exacerbates the hyperventilation-induced hypocapnia during heavy exercise and further lowers both cerebral blood flow and oxygenation. Some indication that cerebral deoxygenation may limit performance comes from the observation that cerebral deoxygenation precedes the development of central fatigue during exercise, which coincides with both reduced cortical motor output and increased cerebral metabolism (Secher et al., 2008; Subudhi et al., 2009; Rasmussen et al., 2010a), and the rapid reversal with an increase in FIO2 mentioned above. Under severe hypoxic conditions, several studies reported a relationship between performance and cerebral deoxygenation during various exercise modes such as repeated sprints (Smith and Billaut, 2010), incremental exercise (Peltonen et al., 2009; Subudhi et al., 2009; Vogiatzis et al., 2011), and static maximal muscle contraction to exhaustion (Rasmussen et al., 2007; Rupp and Perrey, 2009; Goodall et al., 2010). Similarly, exacerbation of the exercise-induced cerebral deoxygenation with nonselective beta-blockade reduces maximal exercise performance under normoxic conditions (Seifert et al., 2009). From these findings, it was hypothesized that reduced cerebral DO2 and associated cerebral tissue deoxygenation may be a limiting factor of performance during exercise in severe hypoxia (Nybo and Rasmussen, 2007; Amann and Dempsey, 2008; Amann and Kayser, 2009).
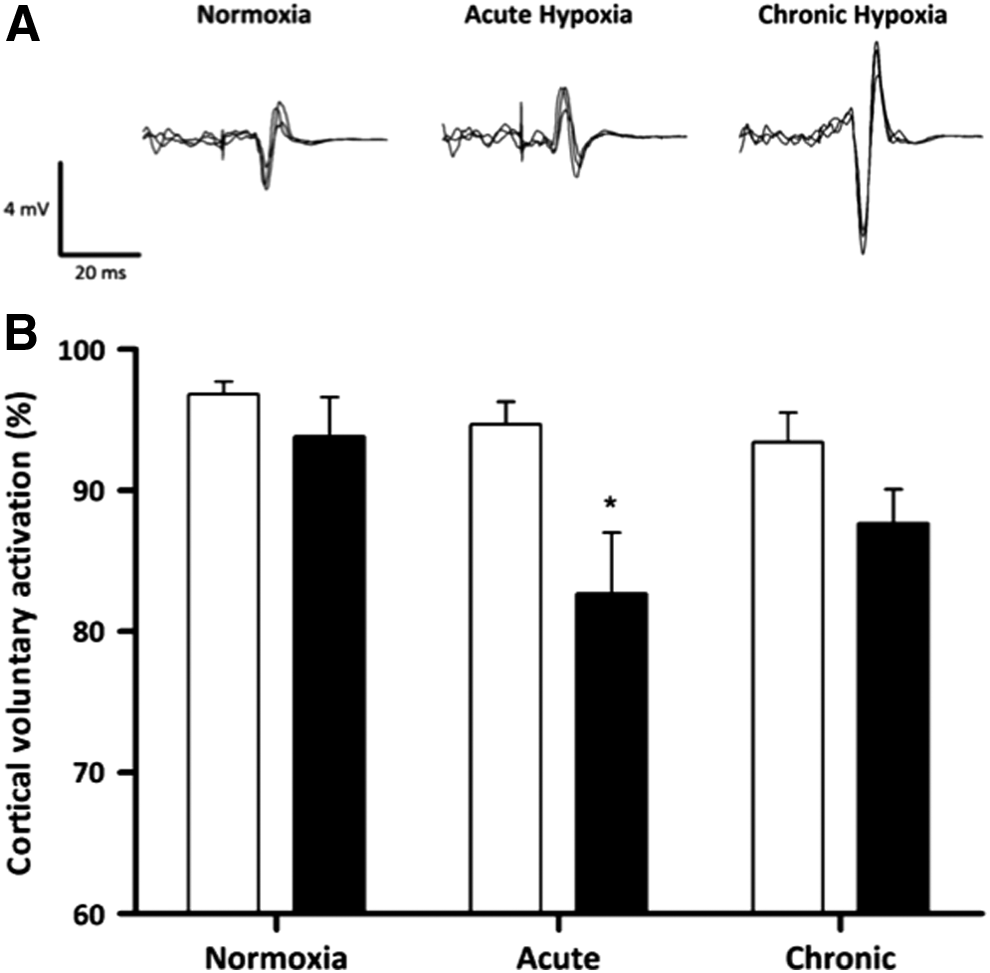
Using complete vascular occlusion of the arm during repeated submaximal isometric elbow flexion to exhaustion, Millet et al. (2012) examined the effect of normoxia and moderate (FIO2: 0.14) and severe hypoxia (FIO2: 0.09) on the development of central fatigue. They found exercise performance was greatly reduced by severe hypoxia compared to moderate hypoxia and normoxia. Since vascular occlusion meant that changes in FIO2 did not affect metabolic condition of the working muscles (as supported by the lack of difference in muscle tissue oxygenation between conditions), they attributed this impaired performance to a direct inhibitory effect of hypoxia on central motor drive, independent of muscle metabolic status. In support of this notion, Goodall et al. (2014) found reduced cortical voluntary activation (i.e., index of supraspinal fatigue) and prefrontal tissue oxygenation (compared to baseline values) following constant-load cycling in acute hypoxia (FIO2: 0.105), while no reductions were observed in normoxia (Fig. 5). Interestingly, these reductions in postexercise cortical voluntary activation and prefrontal tissue oxygenation in acute hypoxia were abolished after 16 days of acclimatizing to 5260 m, while corticospinal excitability was increased twofold compared to pre-exercise values (Fig. 5). The authors concluded that (1) exhaustive exercise in acute hypoxia is associated with the development of central fatigue, which is attenuated after a period of high-altitude acclimatization, and (2) that the observed reduction in central fatigue with acclimatization was likely due to enhanced corticospinal excitability associated with increased cerebral DO2 and tissue oxygenation.
CO2 clamping during dynamic exercise
Like the rapid O2 switch intervention, acclimatization to high altitude is associated with improvement in cerebral DO2, but systemic DO2 also. Therefore, a novel solution was needed to selectively manipulate cerebral DO2 without affecting systemic (i.e., muscle) DO2. CO2 is a potent vasodilator of the cerebral vessels causing global CBF to increase by 2%–4% per mmHg rise in partial pressure of arterial CO2 (Fortune et al., 1992; Sato et al., 2012), while it elicits only small changes in limb blood flow (Lennox and Gibbs, 1932; Ainslie et al., 2005). This unique property suggested that hypercapnia might be an ideal tool to selectively manipulate cerebral DO2 and tissue oxygenation during exercise in severe hypoxia.
Several studies investigated the role of cerebral tissue oxygenation on aerobic capacity by selectively elevating cerebral blood flow (and thereby cerebral DO2) with CO2 breathing during incremental cycling to exhaustion in severe hypoxia (Subudhi et al., 2011; Siebenmann et al., 2013). Subudhi et al. (2011) reported impaired exercise capacity at 1600 m and 4875 m (hypobaric chamber) when they clamped the subjects' partial pressure of end-tidal CO2 (PETCO2) either at (1) 50 mmHg throughout incremental exercise or (2) 40 mmHg from ∼75% maximal power output until exhaustion (Fig. 6). They found ventilation to be greatly elevated (by ∼50 L/min) with PETCO2 clamping during submaximal exercise intensities (37% and 75% of maximal hypoxic power output), compared to room air breathing. Accordingly, they attributed the reduced performance with PETCO2 clamping to earlier functional limitation by the respiratory system with CO2 breathing. Siebenmann et al. (2013) completed those observations by investigating, at a more moderate altitude (3454 m), the impact of clamping PETCO2 at 40 mmHg on aerobic capacity. These studies found clamping PETCO2 increased cerebral blood flow velocity (and presumably cerebral DO2) and attenuated the decrease in cerebral oxygenation, but slightly decreased maximal power output without affecting maximal O2 uptake. Similarly, in altitude naive subjects, we found no improvement in exercise performance during incremental and time trial cycling in severe hypoxia with CO2 breathing (Fan and Kayser, 2013; Fan et al., 2013). During time trial cycling in hypoxia, the modest elevations in both cerebral blood flow velocity and CaO2 with CO2 breathing normalized the cerebral DO2 to normoxic values (Fan et al., 2013). In agreement with findings from Subudhi et al. (2011), we observed no improvement in aerobic capacity or time trial performance with CO2 breathing during exercise in hypoxia. Collectively, those findings do not support the role of reduced cerebral DO2 (and associated cerebral tissue deoxygenation) in limiting performance in hypoxia.
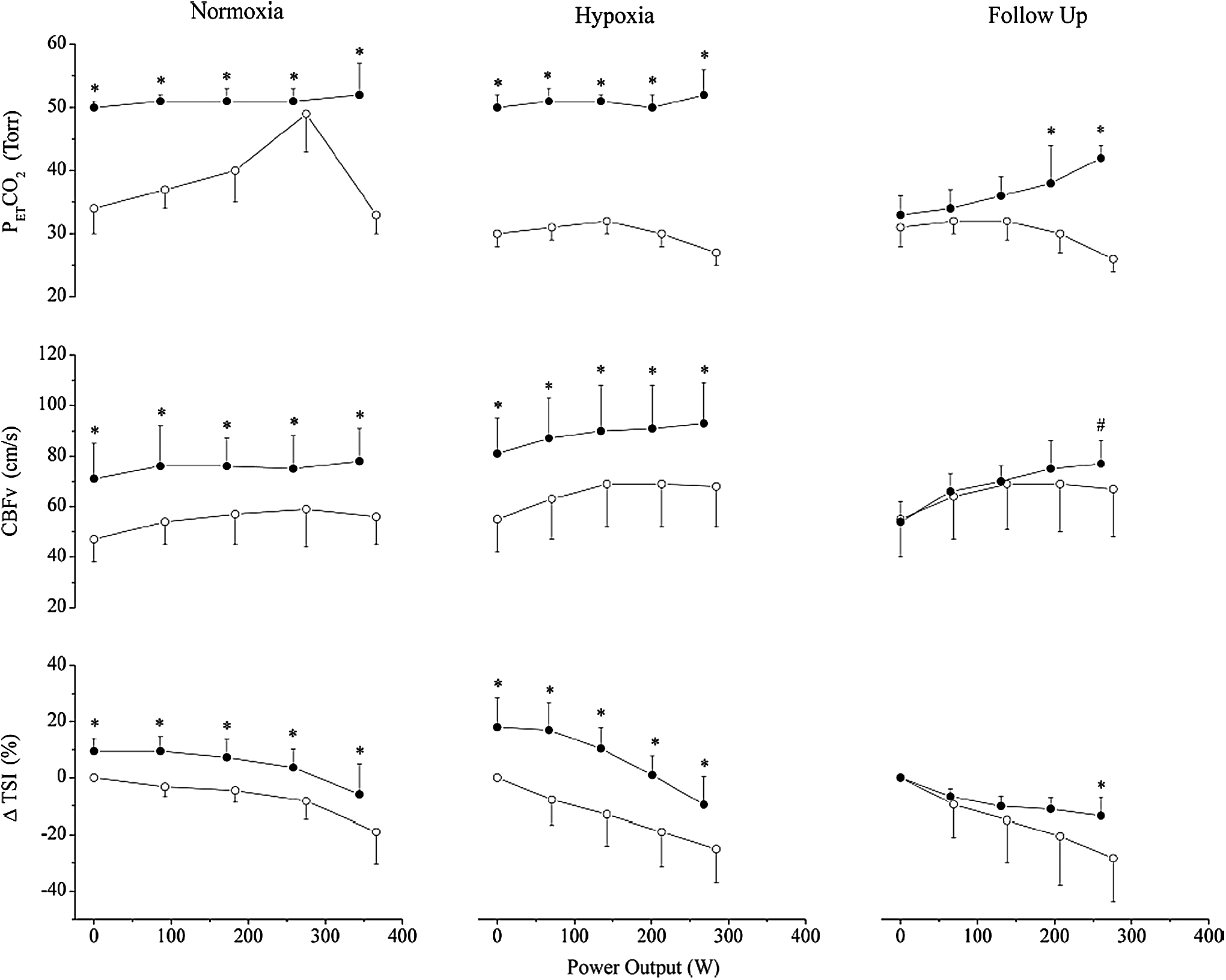
Control (○) and CO2 clamping (•) during incremental exercise in normoxia, hypoxia, and follow-up. Mean ± SD at 0%, 25%, 50%, 75%, and 100% of maximal power output. PETCO2 was clamped at 50 mmHg in normoxia and hypoxia throughout exercise. In follow-up trials, PETCO2 was clamped at 40 mmHg from 75% to 100% maximal power output. Clamping increased cerebral blood flow velocity (CBFv) and cerebral oxygenation, but decreased maximal power output. ΔTSI, change in tissue saturation index. PETCO2, partial pressure of end-tidal CO2. *Different from control (p < 0.05); #nonsignificant trend (p < 0.10). From Subudhi et al. (2011).
The problem with CO2 breathing during exercise
Subudhi et al. (2011) attributed the reduced exercise capacity with PETCO2 clamping in hypoxia in their study to increased respiratory muscle work and associated “steal effect” of blood flow from the working limbs, as well as an elevated rate of perceived exertion (RPE) related to their experimental setup. These factors could have outweighed the benefits of elevated cerebral DO2. They found ventilation to be elevated by ∼50 L/min with PETCO2 clamping during incremental cycling in hypoxia. Likewise, we found ventilation to be elevated by ∼20 L/min with CO2 breathing during incremental and time trial cycling in hypoxia (Fig. 7), while the rate of RPE increase was also higher with CO2 breathing (Fan and Kayser, 2013; Fan et al., 2013). As mentioned before, increased respiratory work exacerbates peripheral fatigue and perception of leg discomfort. Accordingly, the detrimental effects of enhanced ventilatory drive and respiratory work associated with CO2 breathing could outweigh any potential benefits of improved cerebral DO2 and tissue saturation during exercise in hypoxia in our studies.
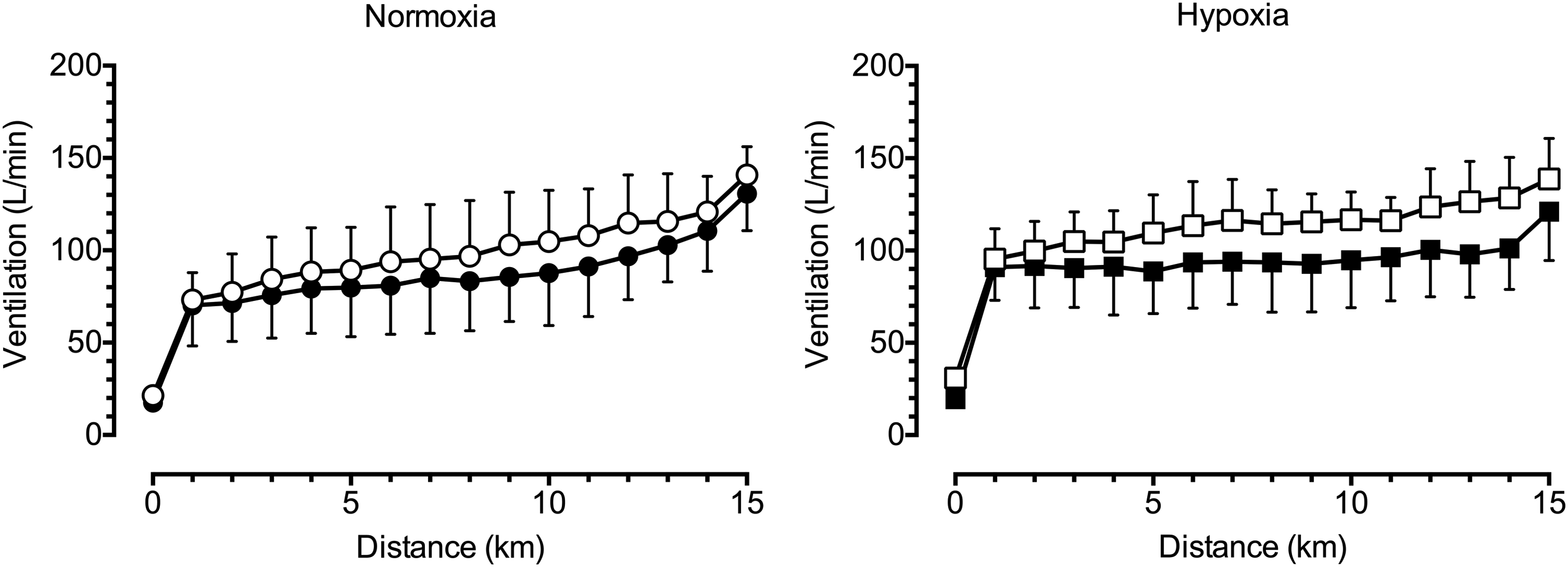
Effect of hypoxia and CO2 clamping on ventilation during 15-km time trial cycling. Both hypoxia and CO2 clamping elevated ventilation during 15-km time trial cycling. Left panel, group data in normoxia (mean ± SD); right panel, group data in hypoxia. •, normoxia control; ◯, normoxia CO2 clamp; ■, hypoxia control; □, hypoxia CO2 clamp. From Fan et al. (2013).
Siebenmann et al. (2013) further argued that CO2 breathing may exacerbate the metabolic acidosis associated with heavy exercise, shifting the oxyhemoglobin curve rightward lowering SaO2, thus limiting maximal O2 consumption. In agreement, we observed a tendency for maximal exercise capacity to be impaired with CO2 breathing in hypoxia (Fan and Kayser, 2013). However, contrary to Siebenmann et al. (2013), we found O2 saturation to be higher with CO2 breathing in hypoxia throughout submaximal exercise intensities, which does not support the notion of a limited pulmonary O2 uptake associated with a rightward shift in the oxyhemoglobin curve.
CO2 clamping during static-limb exercise
During submaximal isometric single knee extension to task failure in hypoxia (peripheral O2 saturation = 80%), Rupp et al. (2015) reported improved prefrontal cortex oxygenation with CO2 clamping, which coincided with improved cortical voluntary activation. Meanwhile, they found CO2 clamping exacerbated peripheral fatigue (as indicated by reduced postexercise index of low-frequency fatigue and torque amplitude) despite elevated tissue oxygenation and blood volume in the working limb during hypoxic exercise. As a result, they did not observe any improvement on performance with CO2 clamping. Since isolated muscle contraction to exhaustion induces moderate cardiorespiratory response in hypoxia (Goodall et al., 2010), data from Rupp et al. (2015) allude to a tantalizing possibility that CO2 clamping might directly augment the development of peripheral fatigue during hypoxic exercise, independent of its respiratory effects. The magnitude and etiology of fatigue depend upon the type and duration of exercise (Enoka and Stuart, 1992), therefore, extrapolating findings from isometric single-limb exercise to dynamic whole-body exercise should be done with caution.
Future Perspectives
Despite the field's best attempts to quantify the changes in cerebral DO2 and cerebral tissue oxygenation during hypoxic exercise, a number of important questions remain unresolved. As Dienel (2013) aptly pointed out, regional heterogeneity and compartmentalization of function and metabolism are one of the hallmark characteristics of the brain. However, the majority of the central fatigue literature treated the brain as a whole (i.e., global DO2 and consumption). The effect of hypoxia on specific brain regions during whole-body exercise remains largely unknown. Existing techniques such as functional MRI, positron emission tomography, electroencephalography, and multisite near-infrared spectroscopy (NIRS) could potentially be used in this area of research. In theory, they would enable the assessment of region-specific cortical and other brain areas' activities. Despite the potential advantages of these techniques, their applications and feasibility in physiological exercise testing have not been well explored, due to the technological and practical limitations as well as the availability and financial costs of these systems. Nevertheless, assessment of neuronal activity could be undertaken with electroencephalography and NIRS, to map out the specific regional changes in cortical activity (and therefore metabolism) during dynamic exercise in severe hypoxia. Findings from these studies would provide the groundwork for future investigations. Quantifying these cortical activity changes during hypoxic exercise would help us identify the specific regions of interest, perhaps even help locate an “origin” of central fatigue in the brain. Some early promising work by Hilty et al. (2011a, 2011b, 2011c) indicates that muscle fatigue leads to changes in interaction between mid/anterior insular, thalamus, and the motor cortex.
A critical consideration in the regulation of exercise performance is whether it is the arterial tissue O2 pressure gradient driving diffusion (i.e., partial pressure of O2 [PO2]), or the blood O2 concentration (i.e., CaO2), which plays a more important role in the development of central fatigue in hypoxia. Increased brain PO2 per se could explain why improved exercise performance was observed with the “O2 switching method,” but not with the “CO2 clamping method,” which increased cerebral DO2 without much of an increase in cerebral PO2. In support, increases in hemoglobin concentration, which restore CaO2 to sea level values, do not restore exercise performance in acclimatized subjects (Calbet et al., 2003). However, the ability to selectively alter brain PO2 poses a significant methodological challenge. Data from traumatic brain injury patients suggest that brain tissue PO2 might be determined by variables related to cerebral O2 diffusion, rather than cerebral DO2 and metabolism (Rosenthal et al., 2008). Whether this is true in healthy populations and how we might measure brain tissue O2 tension in healthy (and intact!) exercising humans present another significant technical issue. It is likely that further advancements of neural imaging techniques for exercise testing are needed before such breakthroughs can be achieved.
Aspects that have been largely overlooked in this field are the psychological and cognitive effects of hypoxia during exercise (Ando et al., 2013). It is conceivable that hypoxia could alter brain processes involved in motivation, both directly and indirectly [e.g., through altered perception of dyspnea (De Peuter et al., 2004; Weinberger and Abu-Hasan, 2009; Aliverti et al., 2011)], which could account for some of the detriments in performance. There is no denying that motivation and mental fatigue play a large part in sporting performances (Marcora et al., 2009; McCormick et al., 2015). Implementing sensitive psychological measurements will improve understanding of the effect of hypoxia on psychological and cognitive functions, thus providing further insight to the mechanisms behind the performance detriments observed during exercise in hypoxia.
Conclusion
There is developing and compelling evidence to suggest that cerebral tissue deoxygenation plays an important role in limiting exercise performance in severe hypoxia. Despite this, recent intervention studies that looked at the effects of an increase in cerebral DO2 and tissue oxygenation with CO2 breathing failed to improve performance during incremental, time trial, and single-limb exercise paradigms. This lack of improvement in performance may be due to an increased ventilatory drive and possibly an augmented peripheral fatigue development associated with hypercapnia during exercise in hypoxia. Novel approaches are clearly needed to tackle this complex matter. Employing alternative neuroimaging techniques may help researchers gain better insights to the “origin” of central fatigue. Identifying the sites involved in central fatigue will bring us one step closer toward clarifying the role of cerebral oxygenation in exercise performance in conditions of reduced oxygen.
Footnotes
Author Disclosure Statement
No competing financial interests exist.