
Abstract
Abstract
Van Iterson, Erik H., Eric M. Snyder, and Bruce D. Johnson. The influence of 17 hours of normobaric hypoxia on parallel adjustments in exhaled nitric oxide and airway function in lowland healthy adults. High Alt Med Biol. 18:1–10, 2017.—Currently, there is a disparate understanding of the role that normobaric hypoxia plays in affecting nitric oxide (NO) measured in exhaled air (eNO) and airway function in lowland healthy adults. Compared to normobaric normoxia, this study aimed to test the effect of 17 hours of normobaric hypoxia on relationships between eNO and airway function in healthy adults. In a crossover study including 2 separate visits, 26 lowland healthy Caucasian adults performed eNO and pulmonary function tests on visit 1 in normobaric normoxia, while repeating all tests on visit 2 following 17 hours of normobaric hypoxia (12.5% O2). Compared to normobaric normoxia, eNO (29 ± 24 vs. 36 ± 28 ppb), forced expiratory volume in one second (FEV1) (4.1 ± 0.7 vs. 4.3 ± 0.8 L), mean forced expiratory flow between 25% and 75% FVC (FEF25–75) (3.9 ± 1.0 vs. 4.2 ± 1.2 L/s), and forced expiratory flow at 75% FVC (FEF75) (2.0 ± 0.7 vs. 2.3 ± 0.8 L/s) increased in normobaric hypoxia, respectively (all p < 0.05). Correlations at normoxia between eNO and FEV1 (r = 0.39 vs. 0.44), FEF25–75 (r = 0.51 vs. 0.51), and FEF75 (r = 0.53 vs. 0.55) persisted as both parameters increased in hypoxia, respectively. For the first time, these data suggest that 17 hours of hypoxic breathing in the absence of low ambient pressure contribute to increased eNO and airway function in lowland healthy adults.
Introduction
I
Originally highlighted in work of in vivo or in vitro animal preparations and subsequently translated to humans, it has been demonstrated that within endothelial, epithelial, and/or inflammatory cells, the indeterminate expression of neuronal, endothelial, or inducible NO synthase (NOS) isoforms may be independently or interactively involved in endogenous NO production within smooth muscle and vessels of lungs (Gustafsson et al., 1991; Hamid et al., 1993; Nijkamp et al., 1993; Asano et al., 1994; Xue et al., 1994; Ward et al., 1995). As such, coinciding with expression of one or more NOS isoforms coupled to complexes of cosubstrates and cofactors (e.g., O2, thiol, etc.), it has been widely suggested that the amount of NO in exhaled air is useful for quantifying pulmonary vascular or airway reactivity evoked by a spectrum of environmental or pharmacologic pulmonary stimuli (Schmetterer et al., 1997; Duplain et al., 2000; Busch et al., 2001; Jones et al., 2001; Verges et al., 2005; Hemmingsson and Linnarsson, 2009; Puckett et al., 2010b; Donnelly et al., 2011; Dweik et al., 2012; MacInnis et al., 2012).
To date, it is recognized that nonspecific lung NO production can be routinely approximated using the fractional exhaled NO (eNO) technique (Dweik et al., 2012; MacInnis et al., 2015). With this, however, it is also apparent that the broad information extractable from eNO in clinical or research settings is not homogeneous, suggesting that both the setting and the study population are critical considerations when interpreting eNO. For example, contrasting interpretation and conclusions drawn from eNO studies in lowland adults during hypoxic breathing suggest that methodological factors involving stimuli delivery (i.e., hypobaria vs. normobaria) may provoke increased, decreased, or no changes in eNO (Verges et al., 2005; Hemmingsson and Linnarsson, 2009; Donnelly et al., 2011; MacInnis et al., 2012; Faiss et al., 2013).
Although the underpinning factor driving varying eNO observations in studies of hypoxia remains unclear, it would appear that changes in eNO stimulated by hypoxia may be sensitive to ambient pressure (normobaric vs. hypobaric) or total time of hypoxic breathing. For example, Faiss et al. (2013) observed that eNO decreased during prolonged (24 hours) hypobaric hypoxia compared to both normobaric normoxia and 24 hours of normobaric hypoxia in lowland healthy adults, whereas eNO in normobaric hypoxia remained invariable compared to baseline normoxia. In contrast, MacInnis et al. (2012) demonstrated that when compared to normoxia, 2–6 hours of normobaric hypoxia provoked increased eNO in lowland healthy adults.
While there is a paucity of evidence suggesting specific directional changes in eNO induced by normobaric hypoxia in healthy adults, to the best of our knowledge, no study has tested effects of normobaric hypoxia on calibrated changes in eNO and airway function in lowland healthy adults. This gap in knowledge is relevant as others have demonstrated inverse relationships between hypoxia-induced changes in eNO and pulmonary systolic arterial pressure, which suggests there may be potential for airway relaxation commensurate with the rise in eNO (Duplain et al., 2000; Busch et al., 2001; Donnelly et al., 2011).
Accordingly, the aim of this study was to assess the influence of normobaric hypoxic breathing on eNO and accompanying changes in airway function in lowland healthy adults. We hypothesized that 17 hours of normobaric hypoxia provokes calibrated increases in eNO and airway function in lowland healthy adults.
Materials and Methods
Participants
Healthy Caucasian adults participated in this study (N = 26, six women). Exclusion criteria included being pregnant, currently smoking or ≥15 pack-year history, or diagnosed with cardiovascular or pulmonary disease as denoted in the Mayo Clinic medical records system and confirmed by the study PI (B.D.J.). All aspects of this study were reviewed and approved by the Mayo Clinic Institutional Review Board. All individuals voluntarily provided written informed consent before participation.
Experimental procedures
All tests were performed during two separate laboratory sessions (normobaric normoxia baseline vs. normobaric hypoxic breathing) separated by >48 hours but not longer than 4 days. Before each study day, participants were asked to abstain from caffeine and alcohol for 12 hours, and avoid exercise and high nitrate/nitrate meals for 48 hours. During initial screening, women of child-bearing age took a pregnancy test, whereas all individuals underwent a blood draw from a vein at the antecubital level to assess levels of Hgb before and immediately at the conclusion of the hypoxic stay.
In addition, at the baseline visit, cardiopulmonary exercise testing (CPET) according to ACSM guidelines was performed by each participant on an upright cycle ergometer to identify any potential cardiopulmonary abnormalities not previously documented in the medical records system for each participant (American College of Sports Medicine, 2013). Involved in this process, participants were asked before CPET whether they complained of shortness of breath coinciding with physical exertion, the study PI (B.D.J.) monitored real-time continuous 12-lead electrocardiogram (ECG) rhythm strips for the presence of test stopping dysrhythmias (e.g., exercise-induced ST-segment depression ≥1.0 mm, multifocal premature ventricular contractions, and sustained ventricular tachycardia), participants were asked whether they stopped exercising because of shortness of breath as opposed to whole body fatigue, and, if present during CPET, participants were asked to report any test terminating symptoms such as moderate to severe angina (American Thoracic Society and American College of Chest Physicians, 2003; American College of Sports Medicine, 2013).
Additionally, objective data relating to breath by breath ventilation and gas exchange (MedGraphics CPX/D; Medical Graphics, Inc., St. Paul, MN) were evaluated real time during CPET as well as post-CPET to identify responses suggestive of abnormal cardiopulmonary function [e.g., inability to achieve >84% predicted peak VO2 (VO2peak) determined from equations of Jones et al. (Jones et al., 1985; American Thoracic Society and American College of Chest Physicians, 2003; American College of Sports Medicine, 2013)]. Identification of whether an individual attained his/her peak effort during CPET was determined by an individual achieving a respiratory exchange ratio ≥1.10 coinciding with a rating of perceived exertion ≥17 (Borg, 6–20 scale) (American Thoracic Society and American College of Chest Physicians, 2003; American College of Sports Medicine, 2013).
At normobaric normoxia (732 ± 2 mm Hg, 21°C, 21% FiO2) during the baseline visit, while in the seated upright position, participants performed baseline eNO and pulmonary function (Medical Graphics CPFS system spirometer; Medical Graphics, Inc.) tests according to ATS guidelines (i.e., flow volume loop: forced vital capacity [FVC], forced expiratory volume in one second [FEV1], mean forced expiratory flow between 25% and 75% of FVC [FEF25–75], FEV1/FVC ratio [calculated], and forced expiratory flow at 75% of FVC [FEF75]) (Miller et al., 2005; Dweik et al., 2012). The diffusing capacity of the lungs for carbon monoxide (DLCO) was performed via standard rebreathe technique as previously described (Snyder et al., 2005). The percent of predicted FVC, FEV1, FEF25–75, and FEV1/FVC was calculated according to equations of Hankinson et al. (1999).
Measurement of eNO
eNO was measured in units of ppb over the course of 10 seconds using a rapid NO analyzer with analysis software provided by Sievers Instruments (GE Instruments, Boulder, CO) using a flow restrictor, allowing for a constant expired flow rate of 50 mL/s against an oral pressure of 5 cm H2O (Dweik et al., 2012). During performance of this test in room air, participants were asked to breathe into the NO analyzer mouthpiece through a filter that minimized room NO contamination. During the hypoxic exposure visit, participants were asked to inspire from a Douglas bag reservoir containing an FiO2 of 12.5%, which permitted testing in the absence of breathing room air. Finally, to help in comparisons with other studies, we also computed eNO converted to units of nm Hg or mPa (MacInnis et al., 2015).
Hypoxic exposure
Participants entered a normobaric (735 ± 2 mm Hg, 21°C) hypoxic tent with controlled inspired oxygen fraction (FiO2 = 12.5%) (Colorado Altitude Training Corporation, Boulder, CO) and were asked to stay in the tent for 17 consecutive hours. Continuous monitoring of heart rate (HR) and oxygen saturation (SaO2) (Nellcor pulse-oximeter; Nellcor, Pleasanton, CA) occurred while in the tent. In addition, every 2 hours a nurse in the Mayo Clinic General Clinic Research Center (GCRC) measured blood pressure (systolic blood pressure [SBP] and diastolic blood pressure [DBP], whereas mean arterial pressure [MAP] was calculated as, DBP plus SBP minus DBP divided by three], hypoxic exposure symptoms (modified Lake Louise scale), and tent gases. At cessation of the hypoxic stay, while remaining in the tent, participants were fit with a facemask that was attached to a large Douglas bag reservoir filled with a gas concentration of 12.5% O2 (also attached to a large portable gas tank consisting of 12.5% O2). This allowed participants to continue to breathe hypoxic air while they were transported via wheelchair to the Integrative Physiological Core Laboratory where repeat tests of pulmonary function and eNO were performed.
Statistical analyses
All parametric data are presented as mean ± standard deviation. Differences between normoxia and hypoxia were computed using two-tailed paired Student's t-tests. To assess the magnitude of influence 17 hours of normobaric hypoxia had on outcomes (e.g., eNO and pulmonary function), power (ideal, ≥0.80) and effect sizes were computed (small = 0.2; medium = 0.5; and large ≥0.8) (Cohen, 1992). Bivariate relationship testing between eNO and pulmonary function was performed using Pearson product moment correlations (correlation coefficient, r) with 95% confidence limits (CL). Correlation coefficients were interpreted as small = 0.10; medium = 0.30; and large ≥0.50 (Cohen, 1992). To ensure differences between conditions were not driven by inordinate eNO data points in either normoxia or hypoxia, subanalyses of data involving eNO and airway function were performed after removal of participant eNO data consistent with observations of MacInnis et al. (2012) as well as potentially lying outside the normal distribution of the mean using tests of Motulsky and Brown (2006). Two-tailed significance was determined using an alpha level set at 0.05. All computations were performed using SAS statistical software v.9.4 (SAS Institute, Cary, NC).
Results
Healthy lowland adults (N = 26) completed both study visits where inspired partial pressure of oxygen (PIO2) was 144 ± 1 versus 86 ± 2 mm Hg in normobaric normoxia versus normobaric hypoxia (p < 0.01), respectively. Participants ranged in age from 21 to 45 years (30 ± 3 years). Basic demographics included a 77%–23% distribution of men to women, respectively, whereas the overall sample height, weight, body mass index, and body surface area were equal to 177 ± 4 cm, 79 ± 5 kg, 25 ± 3 kg/m2, and 2.0 ± 0.1 m2, respectively. Mean VO2peak was 38 ± 3 mL/kg/min, whereas percent predicted VO2peak was 98 ± 8%. At cessation of CPET, respiratory exchange ratio and rate of perceived exertion were 1.16 ± 0.04 and 18 ± 1, respectively.
While there was no clear physiologic or methodologic evidence during testing to explain excessively high eNO [>50 ppb (MacInnis et al., 2012)] in certain healthy participants in this study, in our subanalyses, we excluded data from six individuals who demonstrated eNO >50 ppb at either normoxia or hypoxia. Baseline characteristics from the subset of 20 participants were consistent with those demonstrated in the sample as a whole.
Basic cardiovascular and symptom response to normobaric hypoxia
Table 1 illustrates that 17 hours of normobaric hypoxia resulted in increased HR, SBP, and Lake Louise score compared to normobaric normoxia across the total sample. In contrast, DBP and SaO2 were lower in hypoxia compared to normoxia. Levels of Hgb did not differ between normobaric normoxia and normobaric hypoxia (14.7 ± 1.1 vs. 15.0 ± 1.1 g/dL, respectively; p = 0.14).
Data presented as mean ± SD. N = 26.
SD, standard deviation.
eNO and pulmonary function
Across the entire sample, compared to normobaric normoxia (29 ± 24 ppb, 2.7 ± 2.3 mPa, or 20 ± 17 nm Hg), eNO increased significantly in normobaric hypoxia (36 ± 28 ppb, 3.4 ± 2.7 mPa, or 25 ± 20 nm Hg), which was supported by a large effect size (Fig. 1A, B). This increase in eNO from normoxia (18.9 ± 6.8 ppb, 1.7 ± 0.6 mPa, or 12.9 ± 4.7 nm Hg) to hypoxia (23.4 ± 8.3 ppb, 2.1 ± 0.7 mPa, or 16.1 ± 5.7 nm Hg) was mirrored even following removal of participants demonstrating eNO >50 ppb (Fig. 1C, D).

eNO in normobaric normoxia or 17 hours of normobaric hypoxia.
In contrast, FVC was similar in normoxia (5.2 ± 1.0 L) and hypoxia (5.2 ± 1.0 L) across the total sample (Figs. 2A and 3A) as well as subsample (Figs. 2B and 3B). Consistent with changes in eNO, FEV1 (4.3 ± 0.8 vs. 4.1 ± 0.7 L), FEF25–75 (4.2 ± 1.2 vs. 3.9 ± 1.0 L/s), and FEF75 (2.3 ± 0.8 vs. 2.0 ± 0.7 L/s) were higher in normobaric hypoxia compared to normobaric normoxia, respectively (Figs. 2C–H and 3C–H). These differences were matched by higher FEV1/FVC in hypoxia compared to normoxia in the entire sample (82 ± 3% vs. 79 ± 3%, respectively; p < 0.01; effect size = 0.70, power = 0.93) as well as subsample (81 ± 4% vs. 78 ± 4%, respectively; p < 0.01; effect size = 0.65, power = 0.79). Summarized in Table 2, percent predicted FVC, FEV1, and FEF25–75 were higher in hypoxia compared to normoxia.
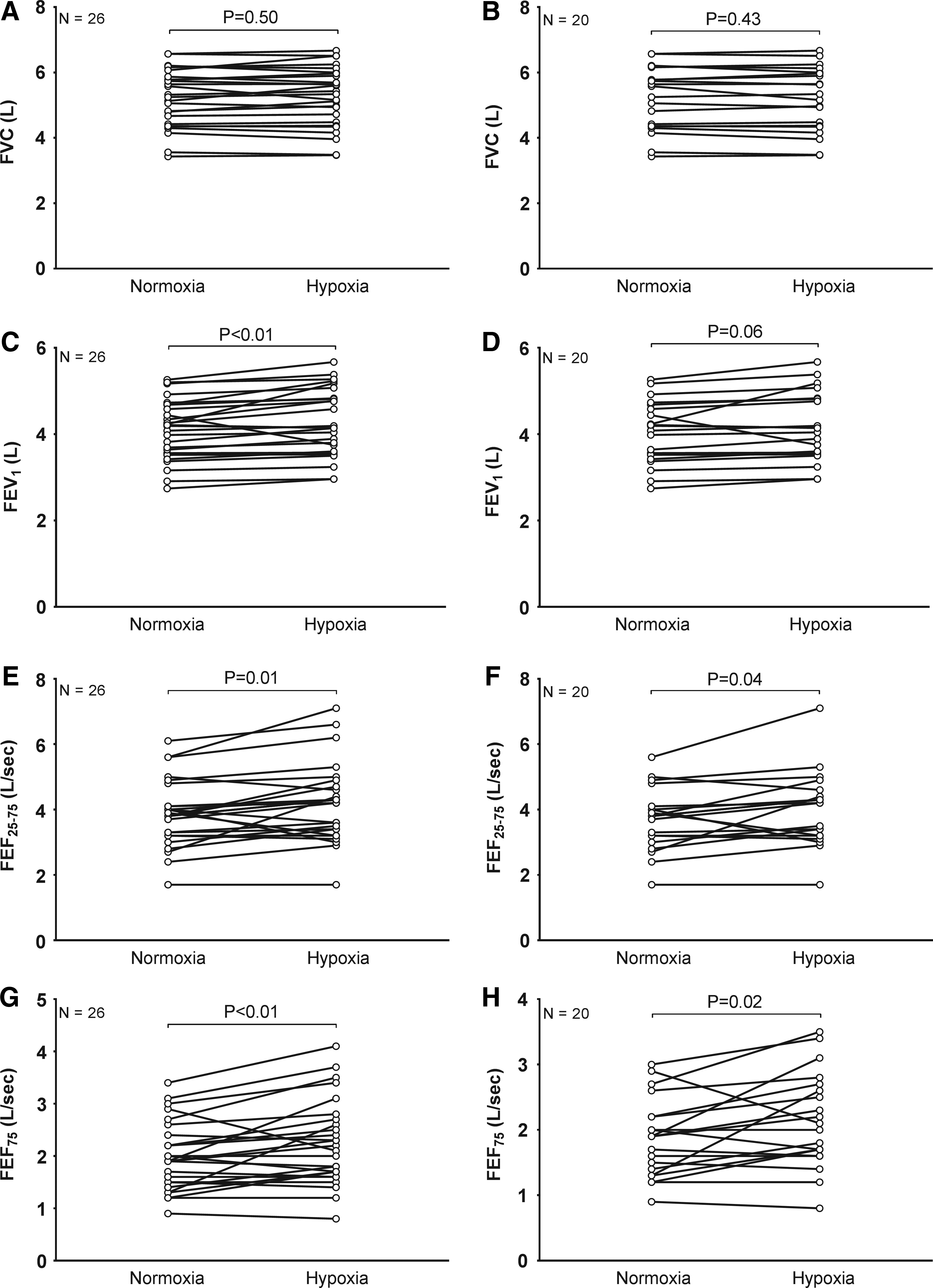
Airway function in normobaric normoxia or 17 hours of normobaric hypoxia.
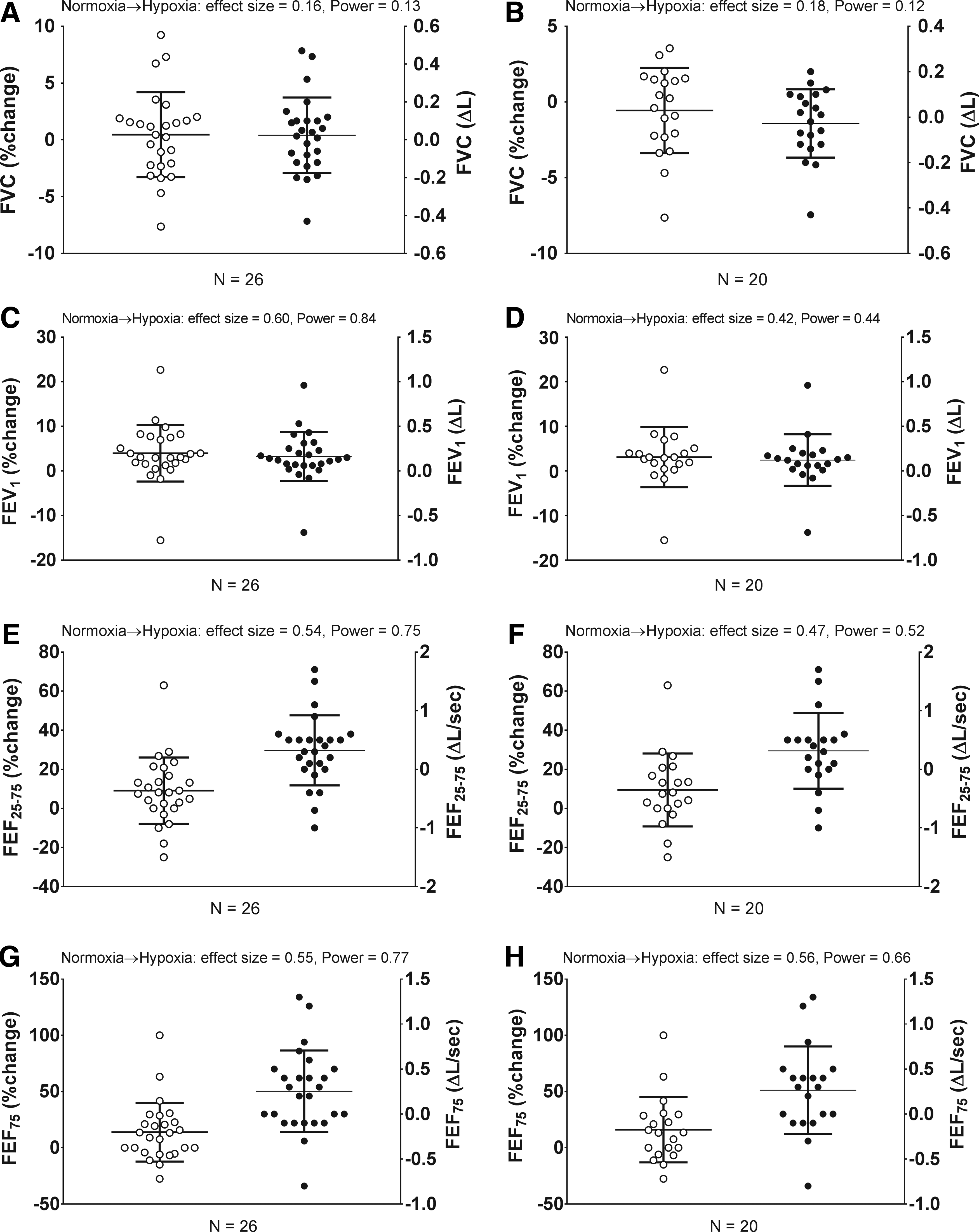
Change in airway function from normobaric normoxia to 17 hours of normobaric hypoxia. Relative % (open circles, left axis) or absolute (Δ, closed circles, right axis) change.
Data presented as mean ± SD or otherwise noted. N = 26. Percent change from normobaric normoxia to normobaric hypoxia (%); absolute change from normobaric normoxia to normobaric hypoxia (Δ). Effect sizes, small = 0.2; medium = 0.5; and large ≥0.8.
FVC, forced vital capacity; FEV1, forced expiratory volume in one second; FEF25–75, mean forced expiratory flow between 25% and 75% of FVC.
For the entire sample, the mean absolute (Δ) increase in DLCO from normoxia [27 ± 3 mL/min/mm Hg] to hypoxia [31 ± 3 mL/min/mm Hg] was 5 ± 2 mL/min/mm Hg (p < 0.01), and the percent increase from normoxia to hypoxia was 15 ± 10%. Increased DLCO from normoxia to hypoxia equated to an effect size of 1.2 at a power of 0.99. These responses were mirrored in the subset of participants (with eNO <55 ppb) in normoxia and hypoxia [27 ± 3 vs. 31 ± 3 mL/min/mm Hg, respectively; p < 0.01; and Δ increase, 5 ± 2 mL/min/mm Hg; percent increase, 14 ± 14%].
eNO relationships
Large positive correlations in Figure 4 were significant in both normoxia and hypoxia for FEV1, FEF25–75, and FEF75, but not for FVC across the total sample, whereas eNO directly correlated with FVC, FEV1, FEF25–75, and FEF75 in individuals demonstrating eNO <55 ppb. Similarly, while eNO directly correlated with DLCO in both normoxia (r = 0.48, 95% CL 0.03 to 0.77, p = 0.03) and hypoxia (r = 0.46, 95% CL 0.00 to 0.75, p = 0.04) in participants demonstrating eNO <55 ppb, there was no correlation between eNO and DLCO in either normoxia (r = 0.12, 95% CL −0.28 to 0.49, p = 0.55) or hypoxia (r = 0.05, 95% CL −0.35 to 0.44, p = 0.80) in the entire sample.
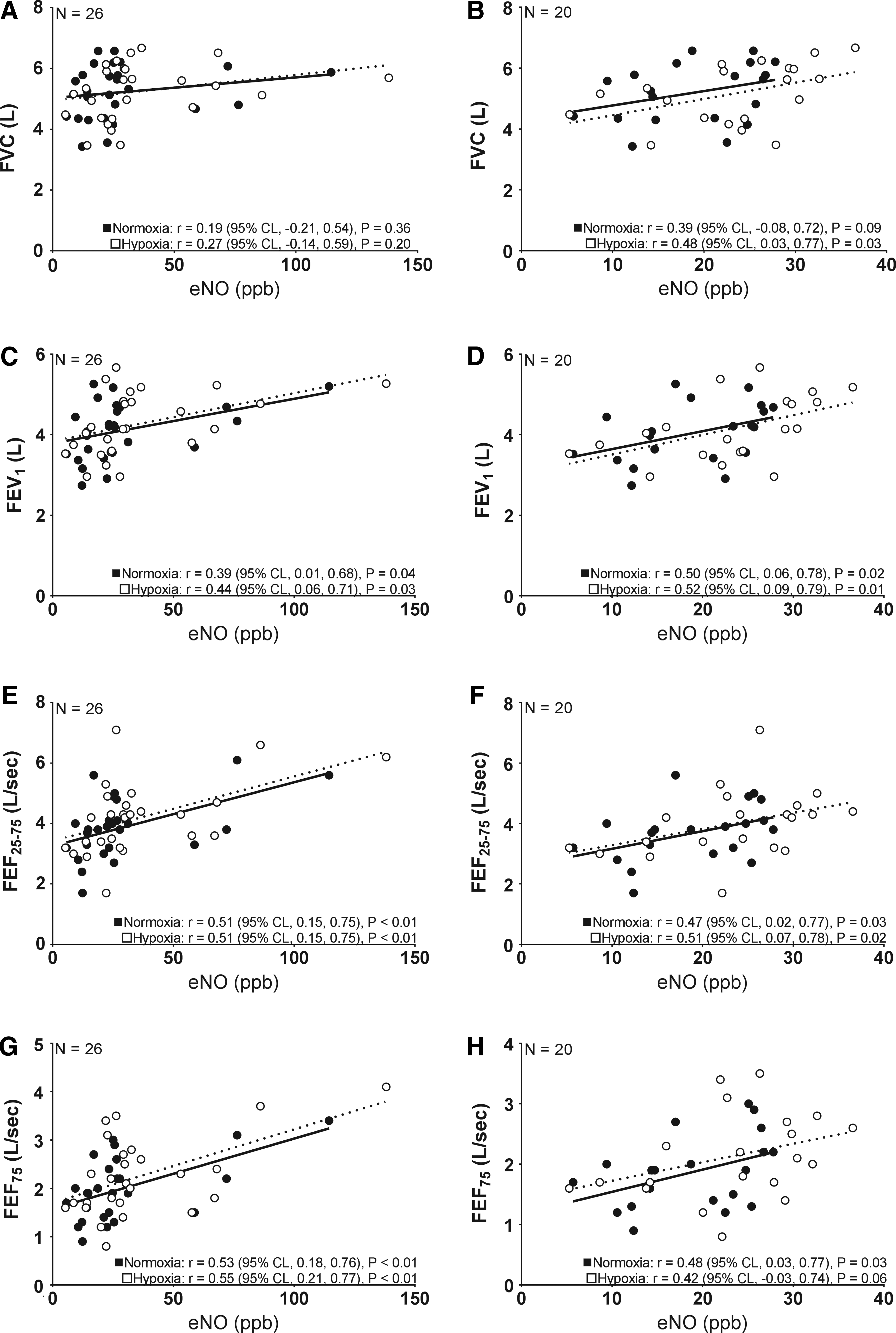
Pearson product moment correlations (correlation coefficient, r, and 95% CL) between eNO and resting airway function at normobaric normoxia (closed circles and solid line) or 17 hours of normobaric hypoxia (open circles and dashed line).
Consistent with absolute FVC, percent predicted FVC did not correlate with eNO in normoxia or hypoxia in the total sample, but trended in the subsample in hypoxia (r = 0.40, 95% CL −0.07 to 0.72, p = 0.07). In neither the total sample nor individuals demonstrating eNO <55 ppb were there significant correlations between eNO and percent predicted FEV1 in normoxia. Whereas both percent predicted FEF25–75 (r = 0.44, 95% CL 0.06 to 0.71, p = 0.03; and r = 0.37, 95% CL −0.10 to 0.70, p = 0.10) and FEV1 (r = 0.29, 95% CL −0.11 to 0.61, p = 0.11; and r = 0.39, 95% CL −0.07 to 0.72, p = 0.08) reached or approached significance in hypoxia for both the total sample and subsample, respectively.
Finally, while patterns were contrasting, relationships between eNO and PIO2 were not significant in either normoxia (r = 0.30, 95% CL −0.17 to 0.65, p = 0.20) or hypoxia (r = −0.22, 95% CL −0.61 to 0.24, p = 0.34) in participants demonstrating eNO <55 ppb, whereas this relationship was significant in normoxia (r = 0.51, 95% CL 0.14 to 0.74, p = 0.01) but not in hypoxia (r = −0.31, 95% CL −0.62 to 0.12, p = 0.15) across the entire sample. eNO did not correlate with SaO2 in either normoxia (r = 0.26, 95% CL −0.15 to 0.58, p = 0.21) or hypoxia (r = −0.18, 95% CL −0.52 to 0.23, p = 0.39) in the total sample, but did so in normoxia (r = 0.56, 95% CL 0.15 to 0.80, p = 0.01) without being matched in hypoxia (r = −0.21, 95% CL −0.60 to 0.25, p = 0.37) for participants demonstrating eNO <55 ppb.
Discussion
The present study demonstrates for the first time that 17 hours of normobaric hypoxia contributes to increased eNO, which is accompanied by augmented airway function in lowland healthy adults.
Hypoxic breathing is associated with numerous changes and adaptations in the pulmonary system that may directly engage bioreactant pathways involving production, transfer, or reactivity of NO. There is controversy, however, as to what extent these adaptations are modulated by stimuli such as hypobaria or normobaria concurrent with hypoxic breathing in lowland healthy adults. In particular, it has been suggested that irrespective of the independent metabolic effect hypoxia might have on lowering NO that has been demonstrated in exhaled air of healthy individuals, more influential to changed (i.e., decreased) eNO is the influence that reduced ambient pressure may have on modulating the chemical properties, bioreactant pathways, or behavior of endogenous NO within lungs and/or systemic plasma circulation (Dweik et al., 1998; Busch et al., 2001; Hemmingsson and Linnarsson, 2009; Donnelly et al., 2011; Faiss et al., 2013).
For example, Hemmingsson and Linnarrson (2009) have ascribed decreased eNO during hypobaric hypoxia to a major influence that low ambient pressure may have on enhancing axial back diffusion of NO into alveoli, which is hypothesized to blunt NO in exhaled air as NO is readily taken up by Hgb-rich pulmonary capillary blood flow (Gibson and Roughton, 1957). Faiss et al. (2013) go on to further suggest that decreased ambient pressure may also influence reduced eNO via its effect on attenuating systemic bioavailability of plasma nitrate and nitrite in hypobaric compared to normobaric hypoxia.
Thus, as noted by Faiss et al. (2013), abnormal changes in systemic plasma nitrate and nitrite may be relevant to eNO because (1) nitrate and nitrite can be actively converted back to NO, (2) NO demonstrates the capacity to diffuse long distances, and (3) the nitrate-nitrite-NO pathway is activated as PIO2 is decreased (Gibson and Roughton, 1957; Lundberg et al., 2008). Nevertheless, while hypotheses of Hemmingsson and Linnarrson (2009) and Faiss et al. (2013) are intriguing discussions attributing blunted eNO to reduced ambient pressure, these data do not support the contention that hypoxia does not play an appreciable role in pathways involved in increasing eNO associated with 17 hours of normobaric hypoxia in lowland healthy adults.
To the best of our knowledge, two studies in addition to ours have observed increased resting eNO evoked by normobaric hypoxia in healthy adults (Verges et al., 2005; MacInnis et al., 2012). Consistent with the present study (i.e., FiO2 = 12.5%), MacInnis et al. (2012) and Verges et al. (2005) demonstrated augmented eNO comparing normoxia to normobaric hypoxia using similar [i.e., FiO2 = 12% (MacInnis et al., 2012)] or more modest levels of hypoxia [i.e., FiO2 = 15% (Verges et al., 2005)]. Interestingly, MacInnis et al. (2012) observed that increased eNO appeared to inversely couple with susceptibility to acute mountain sickness. While we did not test for proneness to acute mountain sickness in this study, MacInnis et al. (2012) demonstrated that individuals without acute mountain sickness had increased eNO levels at baseline persisting throughout normobaric hypoxic exposure (2–6 hours), which was postulated to be the result of favorable effects of high resting arterial oxygen tension (PaO2) and alveolar–arterial oxygen difference on lung NO production. Whereas despite not detailing specific reasons for increased resting eNO comparing normoxia to normobaric hypoxia, it is apparent from observations of Verges et al. (2005) that neither exercise training status (Sheel et al., 2000, 2001) nor predisposition to exercise-induced arterial hypoxemia (related to PaO2) play critical roles in increased eNO associated with acute normobaric hypoxia in otherwise healthy individuals.
In addition to demonstrating increased eNO follows 17 hours of normobaric hypoxia in lowland healthy adults, we also observed these individuals demonstrate augmented airway function directly correlating with eNO. While these relationships are consistent with separate, but related lines of evidence, suggesting inverse links between elevated eNO and pulmonary arterial systolic pressure in various settings of hypoxic breathing in lowland healthy adults (Duplain et al., 2000; Busch et al., 2001; Donnelly et al., 2011), others have contrastingly suggested that both acute hypobaric hypoxia (simulated altitude at 14,000–18,000 ft) and normobaric hypoxia (FiO2 = 10%–12%) lead to reduced FVC in healthy adults (Rahn and Hammond, 1952). Whereas others have similarly observed that hypobaric hypoxia at approximately 17,388 ft (i.e., 5,300 m) leads to reduced FVC and FEV1 in healthy lowland adults, these reductions in airway function are not immediately corrected following administration of supplementary oxygen to increase mean SaO2 from 81 to 94%. (Pollard et al., 1997). Nevertheless, it is important to note that while these data contrast airway function demonstrated in Rahn and Hammond (1952) and Pollard et al. (1997), tests of eNO or systemic bioavailability of NO were not performed by those authors and, therefore, it is difficult to fully appreciate what potential role, or lack thereof, NO production or activity of NO-related bioreactant pathways may have had in influencing reduced airway function at either hypobaric or normobaric hypoxia.
We investigated eNO as a possible indicator of changes in airway NO production stimulated by 17 hours of normobaric hypoxic breathing. Although it has been demonstrated using advanced eNO testing methods that anatomic origins of NO in exhaled air may indeed be localized to upper or lower airways (Tsoukias and George, 1998; Puckett et al., 2010a), both extractable and interpretable data beyond the macroscopic level linked specifically to the direct effect NO has on airway function remain controversial in humans (Hogman et al., 1993). In contrast, bench/animal model work in this line of study suggests that as opposed to NO itself directly causing smooth muscle relaxation and increased airway function, it appears more likely, for example, that NO reactions in the presence of cofactors such as thiol (e.g., forming SNO) and activation of independent or dependent cGMP pathways are more likely involved in contributing to changes in smooth muscle tone (e.g., Ca2+ channel ion sensitivity) leading to airway smooth muscle relaxation and NO in exhaled air (Gaston et al., 1994; Ward et al., 1995; Perkins et al., 1998).
Limitations
We acknowledge that our suggestion of integrated links synonymous with inverse changes in pulmonary vasculature (e.g., ↓ pressure accompanied by, ↑ dilation) with increased eNO and airway function stimulated by 17 hours of normobaric hypoxia is not supported by direct measurements of pulmonary vascular function in this study. Therefore, this is a logical next step in this line of work to bridge this gap in knowledge questioning whether normobaric hypoxia evokes increased eNO commensurate with direct changes in pulmonary vascular hemodynamics and airway function in lowland healthy adults.
This study also did not include serial measurements of eNO and airway function at sequential time increments of normobaric hypoxic breathing. Thus, it is unclear from these data whether there is a duration threshold where exposure to normobaric hypoxia stimulates the strongest changes in eNO and airway function. This may be relevant as others have demonstrated there is no effect of acute hypoxic breathing (e.g., 1–25 minutes) on influencing increased eNO in healthy adults (Schmetterer et al., 1997; Dweik et al., 1998; Donnelly et al., 2011), whereas others have observed augmented eNO within 30 minutes of resting normobaric hypoxia (Verges et al., 2005).
Finally, in addition to the absence of quantifying systemic bioavailability of plasma NO, we did not assess markers of oxidative stress (e.g., reactive oxygen species). The balance of these two factors is suggested to be important as normal bioavailability of plasma NO (i.e., nitrate and nitrite) can contribute to increased NO that may readily react with airway smooth muscle promoting compounds (e.g., thiol) (Gaston et al., 1994; Ward et al., 1995; Perkins et al., 1998; Lundberg et al., 2008), whereas augmented oxidative stress is suggested to degrade NO bioavailability (Thomas et al., 2008). In the context of this study, the potential role of these integrated biomolecular pathways is intriguing as it is suggested that normobaric hypoxia may lead to increased oxidative stress, which would in theory result in decreased bioavailability of plasma NO and, hence, attenuated airway relaxation, airway function, and eNO (Thomas et al., 2008; Pialoux et al., 2009). A deeper understanding of the influence normobaric hypoxia has on oxidative stress and NO formation or metabolism is warranted to understand why, if present in this study, airway function increased despite potential effects of hypoxia-mediated oxidative stress on NO pathways.
Conclusions
Compared to normobaric normoxia, these data suggest that 17 hours of normobaric hypoxia stimulates calibrated increases in eNO and airway function in lowland healthy adults. Augmented eNO and airway function resulting from 17 hours of hypoxia cannot be attributed to the effects of low ambient pressure as all measurements occurred at sea level. More elaborate and sensitive secondary studies assessing the potential association between normobaric hypoxia and NO reactions with molecules or compounds that are linked to changes in airway smooth muscle tone are needed to better understand the full implications of these data.
Footnotes
Acknowledgments
We thank Kathy O'Malley, Angela Tarara, and Minelle Hulsebus for their help with data collection. We would like to thank the staff of the General Clinical Research Center (GCRC) for their assistance throughout this study. The Mayo Clinic GCRC is supported by the U.S. Public Health Service Grant M01-RR00585. This work was supported by the NIH Grant HL71478 (B.D.J.) and AHA Grant 16POST30260021 (E.V.I.).
Author Disclosure Statement
The authors and/or study team members involved in this study do not have any competing financial interests to disclose.