
Abstract
Abstract
Background:
This study aimed to explore the effects of netrin-1 on hypobaric hypoxia-induced lung injury in mice.
Methods:
We exposed 6–8-week-old C57BL/6 mice to hypobaric stress at 340 mmHg for 30 minutes followed by 260 mmHg for different periods (6, 12, 18, and 24 hours) to observe the severity of lung injury (O2 concentration, 21%; 54.6 mmHg). The wet/dry weight ratio and protein leakage from the mouse lung were used to determine the suitable exposure time. Netrin-1 was injected into the tail vein of mice before 18-hour decompression. Inflammatory cytokines, lung injury scores, and activity of nuclear factor κB were evaluated. The expression of apoptosis-related proteins was also examined.
Results:
Protein concentration in the bronchoalveolar lavage fluid was significantly higher in the 18-hour group (p < 0.05). Pulmonary pathology revealed neutrophil infiltration, alveolar septum thickening, and tissue edema. Injury score and macrophage inflammatory protein 2 levels were also increased. Intrinsic apoptosis pathway was activated. Hypoxia decreased the expression of Bcl2 protein, the number of active caspase-3-stained cells, and UNC5HB receptors. Pretreatment with netrin-1 reduced protein leakage, inhibited neutrophil migration, lowered the injury score, attenuated apoptosis, and increased UNC5HB receptor expression.
Conclusion:
Netrin-1 dampens hypobaric hypoxia-induced lung injury by inhibiting neutrophil migration and attenuating apoptosis.
Introduction
Hypobaric hypoxia occurs at a high altitude due to low atmospheric pressure and low partial pressure of oxygen, which leads to a stress condition that deprives the body of a normal supply of oxygen (Pandey et al., 2017). Under such conditions, uneven hypoxic pulmonary vasoconstriction occurs, given that both pulmonary artery and vein pressure increases. The microvascular pressure of the pulmonary capillary beds also increases, which in turn induces stress failure. The disruption of capillary bed causes leakage in the pulmonary blood–gas barrier. Besides, impaired clearance of alveolar fluid facilitates the accumulation of edema fluid in the lung (Swenson et al., 2002). Inflammation of the respiratory tract, including the alveolar space, also contributes to edema formation at lower capillary pressures (Bartsch et al., 2005).
Netrin-1, first discovered during the 1990s, is a neuronal guidance protein. It is a laminin-related molecule secreted at the spinal cord midline whose function is to guide vertebrate commissural axons (Kennedy et al., 1994; Serafini et al., 1994). Recently, the immunomodulatory function of netrin-1 was described (Mirakaj and Rosenberger, 2017). In addition to its potential of inhibiting leukocyte migration at the endothelial interface (Ly et al., 2005), netrin-1 also regulates the inflammatory responses of neutrophils and macrophages (Ranganathan et al., 2013). Previous studies have demonstrated that netrin-1 attenuates hypoxia-elicited inflammation at mucosal surfaces and significantly dampens the extent of acute lung injury (Rosenberger et al., 2009; Mirakaj et al., 2010). In neuronal tissues, netrin-1 has recently been shown to reduce neuronal injury by attenuating neuronal apoptosis (Chen et al., 2017; Xie et al., 2018).
Most of the previous experiments on acute lung injury induced by hypobaric hypoxia were conducted on rats, and experiments on mice were rare (Tan et al., 2015). Particularly, the results based on the different experimental protocols for studies on mice were inconsistent. The potential of netrin-1 in preventing hypobaric hypoxia-induced lung injury deserves further investigation. In this study, we attempted to establish a mouse model of acute lung injury induced by hypobaric hypoxia and examined the activity of netrin-1.
Materials and Methods
Animal preparation and the hypobaric chamber
The use of animals in this study conformed to the National Institutes of Health guidelines (National Academy Press, 1996), and approval of the project protocol was obtained from the National Science Council and Animal Review Committee of the National Defense Medical Center.
Six to 8-week-old male C57BL/6 mice (each weighing 15–25 g) were used (BioLASCO Co., Ltd., Taiwan). During the initial 1-week period of acclimation, mice were maintained in the animal center at 23°C ± 1°C with a 12-hour light/dark cycle. Access to clean food and water was unrestricted.
The hypobaric chamber was self-designed (Lacidem Vacuum Machine 3; Lacidem International Co., Ltd., Taipei, Taiwan).
Experimental protocol of hypobaric hypoxia exposure
A total of 30 mice were randomized into five equal groups. One group served as the control; 6-, 12-, 18-, and 24-hour groups were exposed to hypobaric hypoxia for 6, 12, 18, and 24 hours, respectively. The exposure period was designed based on the results of Sarada et al. (2008). Hypobaric exposure was divided into two consecutive phases, namely, adaptation phase and experimental phase. During the adaptation phase, mice were exposed to 340 mmHg (equivalent to ∼6250 m) pressure in a hypobaric chamber for 30 minutes. During the experimental phase, mice were exposed to 260 mmHg (equivalent to ∼8230 m) pressure in a hypobaric chamber for different prescheduled periods.
Circulation of fresh air and carbon dioxide venting was maintained via a regulating valve. Access to clean food and water was unrestricted.
Experimental protocol of netrin-1 pretreatment
Mice in the vehicle group were pretreated with 100 μL phosphate-buffered saline (PBS) containing 0.1% bovine serum albumin (BSA), via tail vein injection (Sigma Chemical Co., St. Louis, MO). Mice belonging to the netrin-1 group were pretreated with 500 ng netrin-1 (Recombinant Chicken Netrin-1 Protein; R&D Systems, Inc., Minneapolis, MN) in 100 μL PBS containing 0.1% BSA, via tail vein injection. The mice in both groups were exposed to 340 mmHg for 30 minutes followed by exposure to 260 mmHg in the hypobaric chamber. The period of experimental phase was designed according to the results of previous studies involving hypobaric hypoxia exposure.
Mice were anesthetized with intraperitoneal sodium pentobarbital (60 mg/kg b.w.; SCI Pharmtech, Inc., Taipei, Taiwan) and sacrificed after hypobaric hypoxia exposure.
Measurement of lung wet/dry weight ratios
The left lung lobe was removed from the hilar region, and wet weight was determined. Then, it was placed in an oven at 60°C for 48 hours to determine the wet/dry weight ratio.
Protein concentration in the bronchoalveolar lavage fluid
After a median sternotomy and tracheostomy, bronchoalveolar lavage fluid was obtained by irrigating the right lung twice with 0.7 mL saline from tracheostomy tube. The fluid was centrifuged at 200 g for 10 minutes at 4°C. The supernatant was collected and stored at −75°C until further use. The concentration of protein in the supernatant was measured using the bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL).
Measurement of tumor necrosis factor-α and macrophage inflammatory protein 2
Blood sample was obtained from the heart and then centrifuged at 2000 g for 20 minutes at 4°C. The plasma was collected and stored in the refrigerator at −75°C until further use. The plasma levels of tumor necrosis factor-α (TNF-α) and macrophage inflammatory protein 2 (MIP-2) were measured using the ELISA kit (R&D Systems, Inc., Minneapolis, MN).
Histological analysis
The left lung lobes were expanded by instillation of 10% formaldehyde solution at a hydrostatic pressure of 20 cmH2O from tracheostomy tube and fixed in 10% formaldehyde for 24 hours. The specimens were sent to Taipei Institute of Pathology for further management. The lungs were stained with hematoxylin and eosin. The number of polymorphonuclear neutrophils in the lung interstitium was evaluated as the average number of polymorphonuclear neutrophils per high-power field (400 × ). For each section, a minimum of 10 fields were randomly examined by an inspector unaware of the protocol, and the extent of lung injury for each field was scored according to (1) the infiltration or aggregation of neutrophils in the airspace or vessel wall, and (2) the thickness of the alveolar wall. Each assessment was graded 0, 1, 2, or 3, for no, mild, moderate, or severe injury, respectively. The resulting two scores were added up and presented as the lung injury score for that section (Wu et al., 2013).
Western blot analysis
Cytoplasmic and nuclear proteins were extracted from frozen lung tissue using the Nuclear/Cytosol Extraction kit (BioVision, Inc., Mountain View, CA) according to the manufacturer's instructions. Protein concentrations were measured using the BCA protein assay (Pierce, Rockford, IL). Equal amounts of lung homogenates (30 μg per lane) were fractionated on 10%–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels, and transferred to Hybond polyvinylidene fluoride membranes (Sigma Chemical Co.). The membranes were blocked by incubation in PBS containing 0.1% Tween 20 (PBST) and 5% nonfat milk for 1 hour at room temperature. Blots were incubated with B-cell lymphoma 2 (Bcl-2) and anti-nuclear factor κB (NF-κB) p65 polyclonal antibodies (diluted 1:1000; Cell Signaling Technology, Danvers, MA) overnight at 4°C. The blots were then washed in PBST three times for 10 minutes. Next, the blots were incubated with horseradish peroxidase-linked anti-rabbit IgG (1:40,000) or anti-mouse IgG (1:50,000) for 1 hour at room temperature, followed by washing thrice in PBST for 10 minutes each. Bands were visualized using enhanced chemiluminescence reagents and by exposing the blot to X-ray film. The blots were then stripped and incubated with either anti-TATA antibody (for nuclear protein, diluted 1:1000; Abcam, Cambridge, MA) or anti-β-actin antibody (for cytoplasmic protein, diluted 1:10,000; Sigma Chemical Co.) to ensure equal loading (Wu et al., 2013).
Activated caspase-3 immunohistochemistry
Formalin-fixed paraffin sections (4 μm) were de-paraffinized before antigen retrieval, and endogenous peroxidase was blocked using 3% hydroxy peroxide in methanol for 15 minutes. The slides were then incubated for 60 minutes with a polyclonal antibody (1:200 dilution; Cell Signaling Technology, Beverly, MA) that specifically recognizes the large fragment (17/19 kD) of activated, but not full length, caspase-3. After washing, slides were sequentially incubated with rat tissue-specific horseradish peroxidase-polymer anti-rabbit antibody (Nichirei Corporation, Tokyo, Japan) for 30 minutes. Horseradish peroxidase was then reacted with diaminobenzidine substrate for 3 minutes, followed by counterstaining of sections with hematoxylin (Wu et al., 2013).
Immunofluorescence staining for netrin-1 and UNC5HB
Rabbit polyclonal antibodies to netrin-1 and UNC5H2 (diluted 1:100; Bioss Antibodies, Inc., Woburn, MA) were used for immunofluorescent labeling. The labeled netrin-1 antibody was incubated for 30 minutes with chicken anti-rabbit IgG-FITC (green) (diluted 1:100; Santa Cruz Biotechnology, Dallas, TX) at room temperature. Goat anti-rabbit IgG-PE (red) (diluted 1:100; Santa Cruz Biotechnology) was used as the secondary antibody to the labeled UNC5H2 antibody. Images were collected using a fluorescence microscope (Leica DM 2500; Wetzlar, Germany) (Liao et al., 2017).
Statistical analysis
Data are expressed as mean ± SD. Statistical differences between group means were determined with Kruskal-Wallis test, followed by a post hoc comparison using the Mann-Whitney U test. Significance was determined at p < 0.05.
Results
Wet/dry weight ratio
The wet/dry weight ratios were not significantly different between control and other groups.
Lung lavage protein concentration
The lung lavage protein concentration was significantly higher in the 18-hour group than in the control group (Fig. 1A; p < 0.05), and this increase in concentration was attenuated upon netrin-1 pretreatment (Fig. 1B; p < 0.05).

Bicinchoninic acid protein assay of BALF.
Pathological findings and injury score
Microscopic examination revealed perivascular edema and higher inflammatory cell infiltration in the 18-hour group than in the control group (Fig. 2A). Netrin-1 pretreatment decreased neutrophil infiltration (Fig. 2A, B) and lung injury scores (Fig. 2C) (p < 0.05).
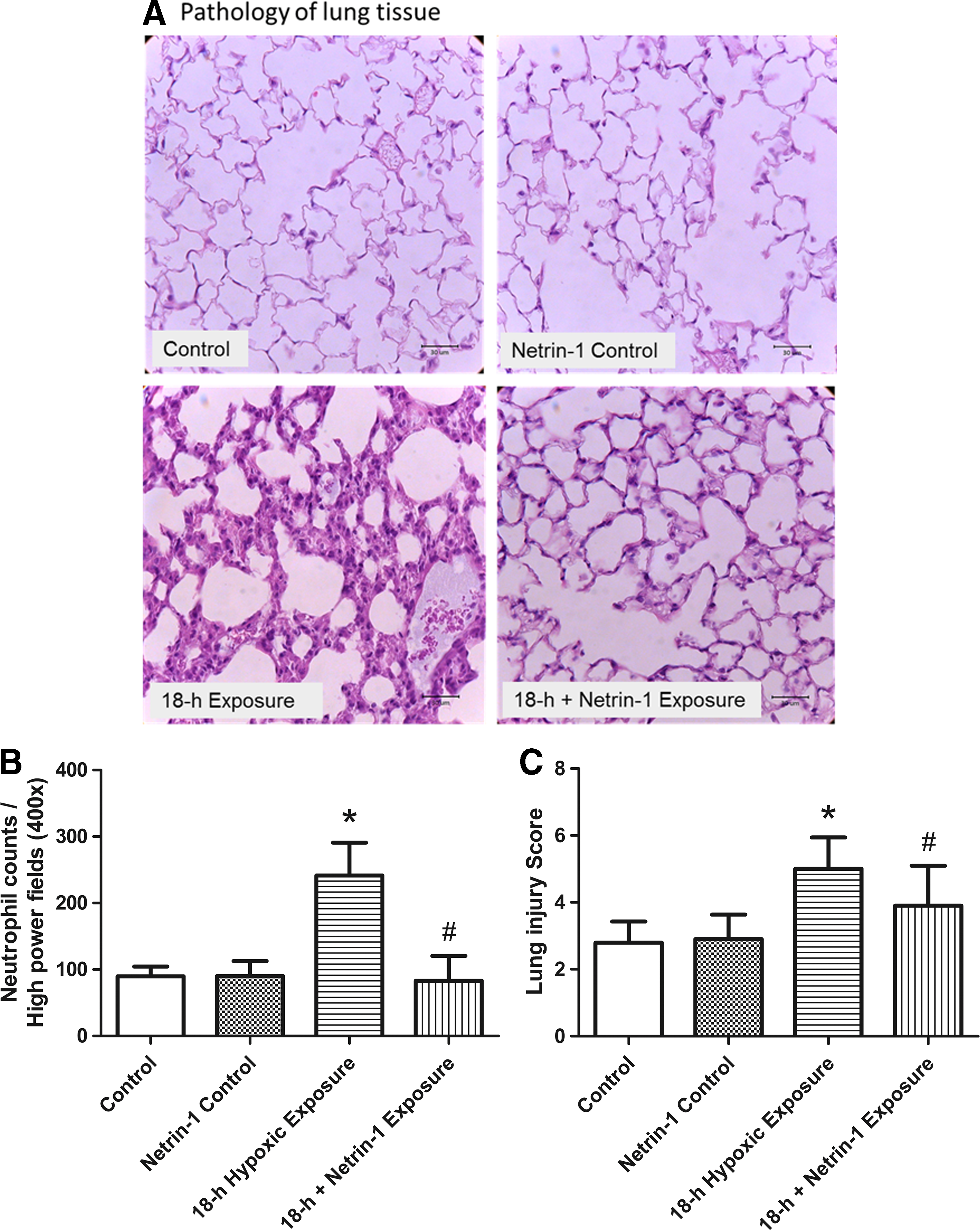
Pathology of lung tissue and lung injury scores. As shown by a representative micrograph of lung tissue (400 × magnification),
TNF-α production and MIP-2 in plasma
The TNF-α level in the plasma was undetectable.
The plasma MIP-2 level (Fig. 3) was significantly higher in the 18-hour group than in the control group (p < 0.05). MIP-2 elevation was attenuated upon pretreatment with netrin-1 (p < 0.05).
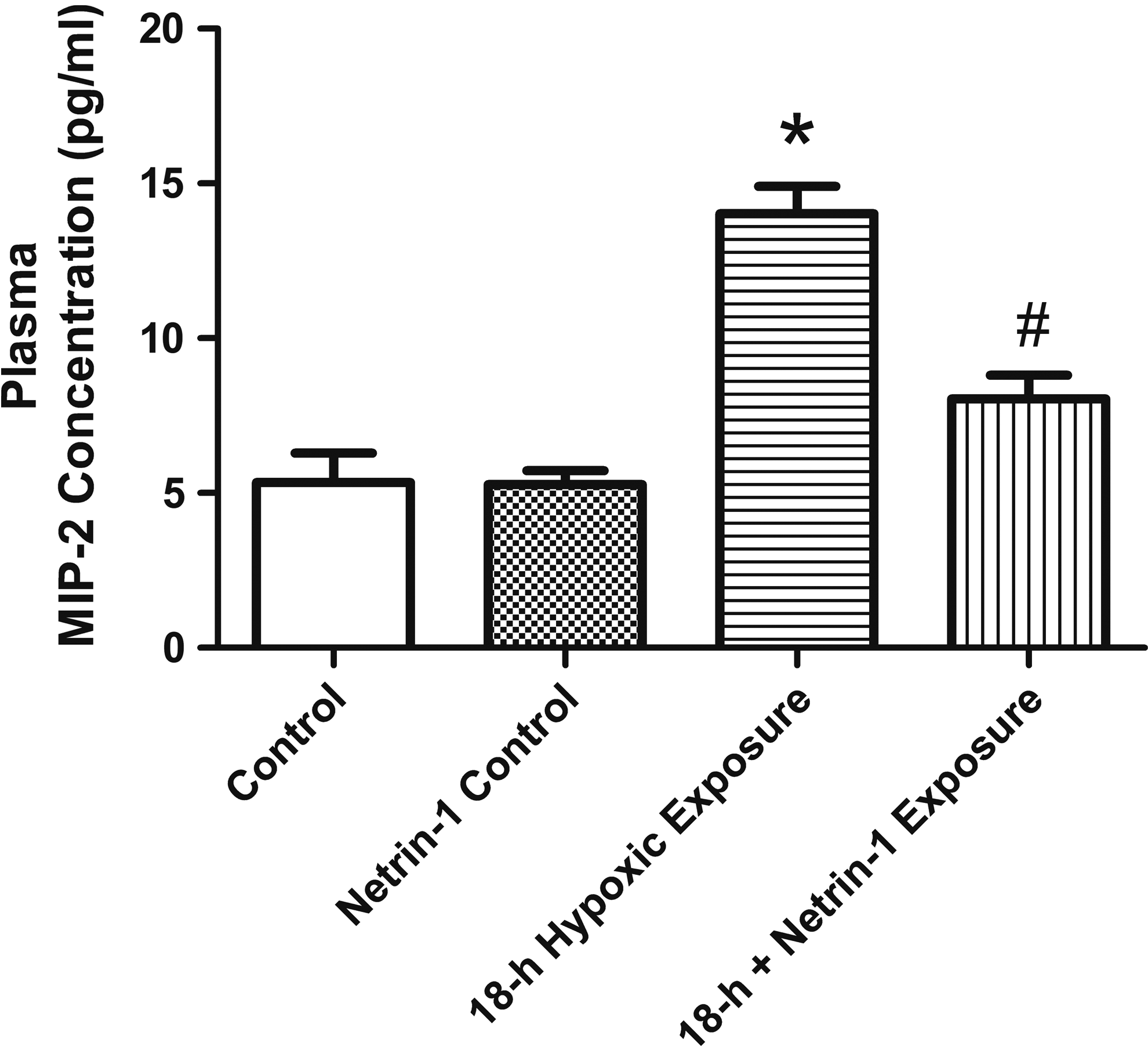
Plasma concentration of MIP-2. Plasma MIP-2 levels increased significantly in the 18-hour group, which were significantly attenuated by pretreatment with netrin-1. Data are expressed as mean ± SD (n = 5). *Significantly different from the control group (p < 0.05); #significantly different from the 18-hour group (p < 0.05). MIP-2, macrophage inflammatory protein 2.
NF-κB signaling pathway
The nuclear level of phospho-NF-κB p65 was significantly higher in the 18-hour group than in the control group. There was no significant difference between the 18-hour group with or without netrin-1 pretreatment (Fig. 4).
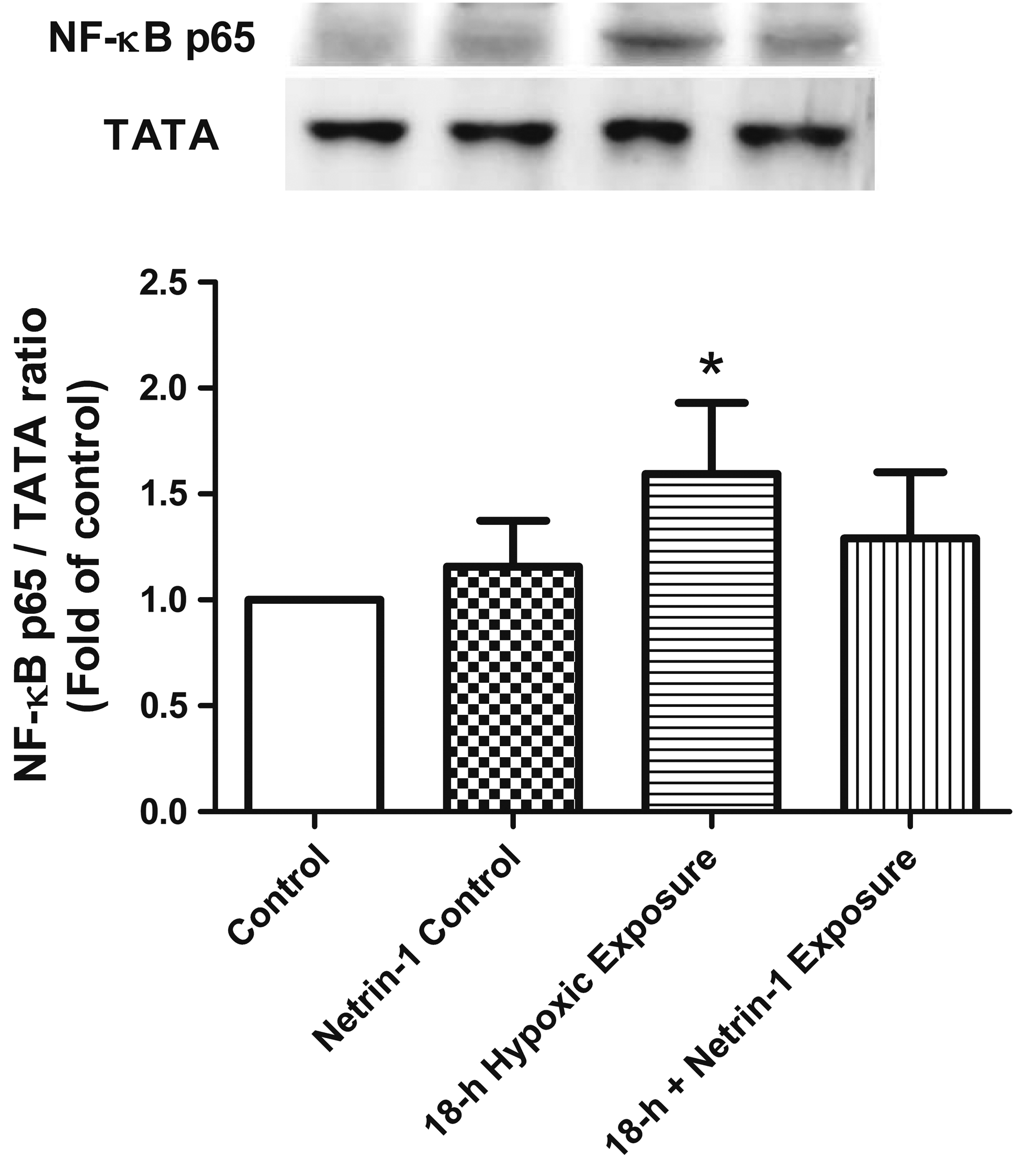
Western blotting of NF-κB. The level of nuclear phospho-NF-κB p65 was significantly higher in the 18-hour group than in the control group. TATA served as a loading control for nuclear proteins. A representative blot is shown. Hypobaric hypoxia upregulated NF-κB p65 protein expression. Data are expressed as mean ± SD (n = 3). *Significantly different from the control group (p < 0.05). NF-κB, nuclear factor kappa-B.
Apoptosis
Compared with the control group, the 18-hour group exhibited a significantly lower level of Bcl-2 protein in the lung tissue, which was attenuated upon netrin-1 pretreatment (Fig. 5A).
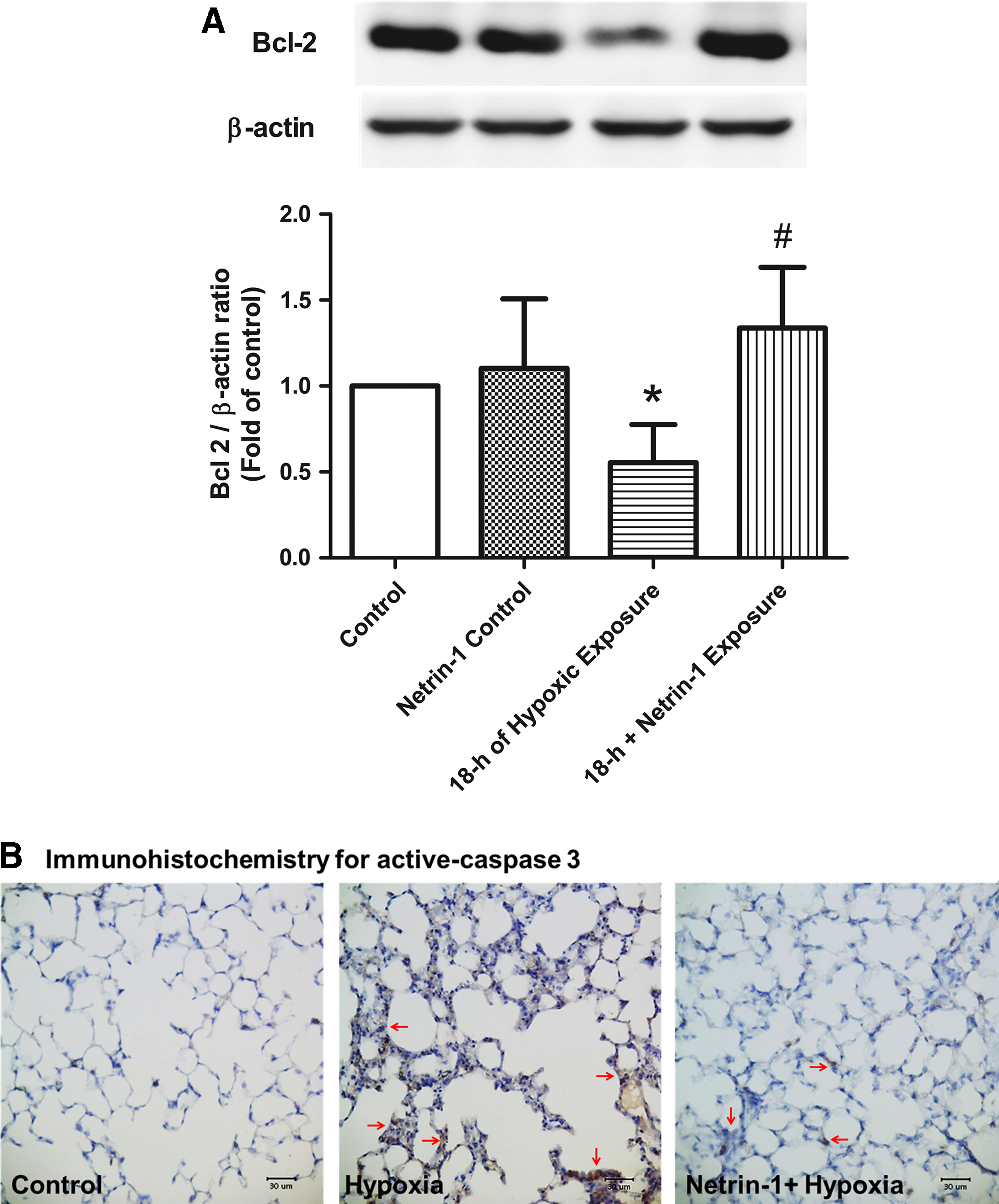
Protein expression involved in the apoptosis pathway.
The density of activated caspase-3-immunolabeled cells was significantly higher in the 18-hour group than in the control group. Pretreatment with netrin-1 significantly decreased the abundance of immunolabeled cells (Fig. 5B).
Expression of netrin-1 receptor UNC5HB
Compared with the control group, the 18-hour group showed slightly increased netrin-1 protein expression. Pretreatment with netrin-1 increased netrin-1 protein expression in both netrin-1 control group and 18-hour plus netrin-1 exposure group (Fig. 6A).
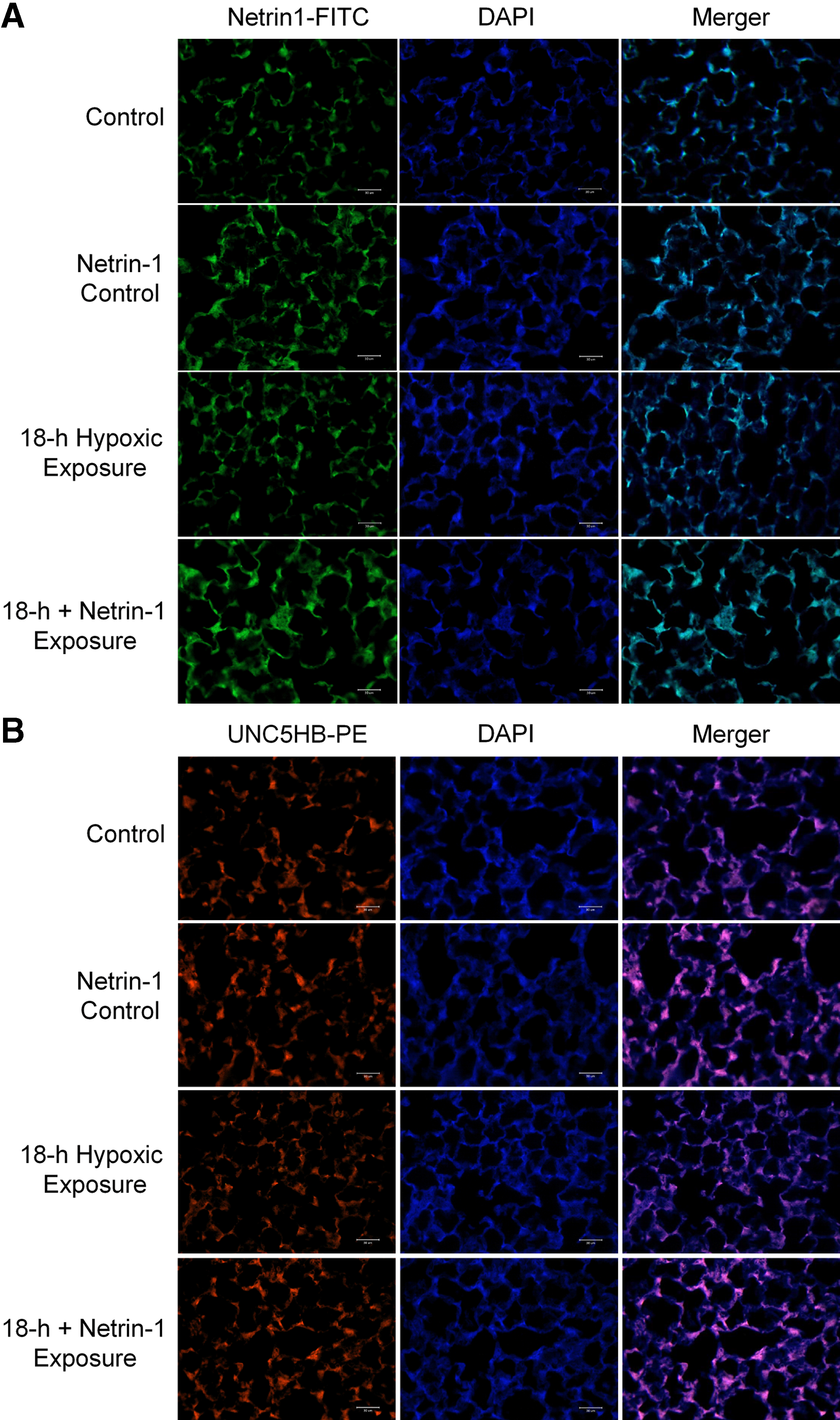
Compared with the control group, the 18-hour group showed decreased netrin-1 receptor UNC5HB expression. Pretreatment with netrin-1 increased netrin-1 receptor UNC5HB expression in netrin-1 control group. Netrin-1 pretreatment reversed UNC5HB downregulation induced by 18-hour hypoxic exposure (Fig. 6B).
Discussion
The purpose of this pilot study was to establish an experimental model of acute lung injury induced upon hypobaric hypoxia in mice and to investigate the prophylactic effects of netrin-1. Mice belonging to the 18-hour group (18-hour exposure to 260 mmHg) exhibited lung injury. Although there was no obvious pulmonary edema, apoptosis occurred in combination with elevated neutrophil counts and plasma MIP-2 levels. Under such circumstances, netrin-1 could inhibit neutrophil migration and reduce MIP-2 secretion and protein leakage. Apoptosis was attenuated, so acute lung injury induced upon hypobaric hypoxia could be dampened. The protective effect of netrin-1 might be through UNC5HB receptor. A flow diagram of our experimental protocol is shown in Figure 7. To our knowledge, this is the first study that explores the effects of netrin-1 on acute lung injury induced upon hypobaric hypoxia.
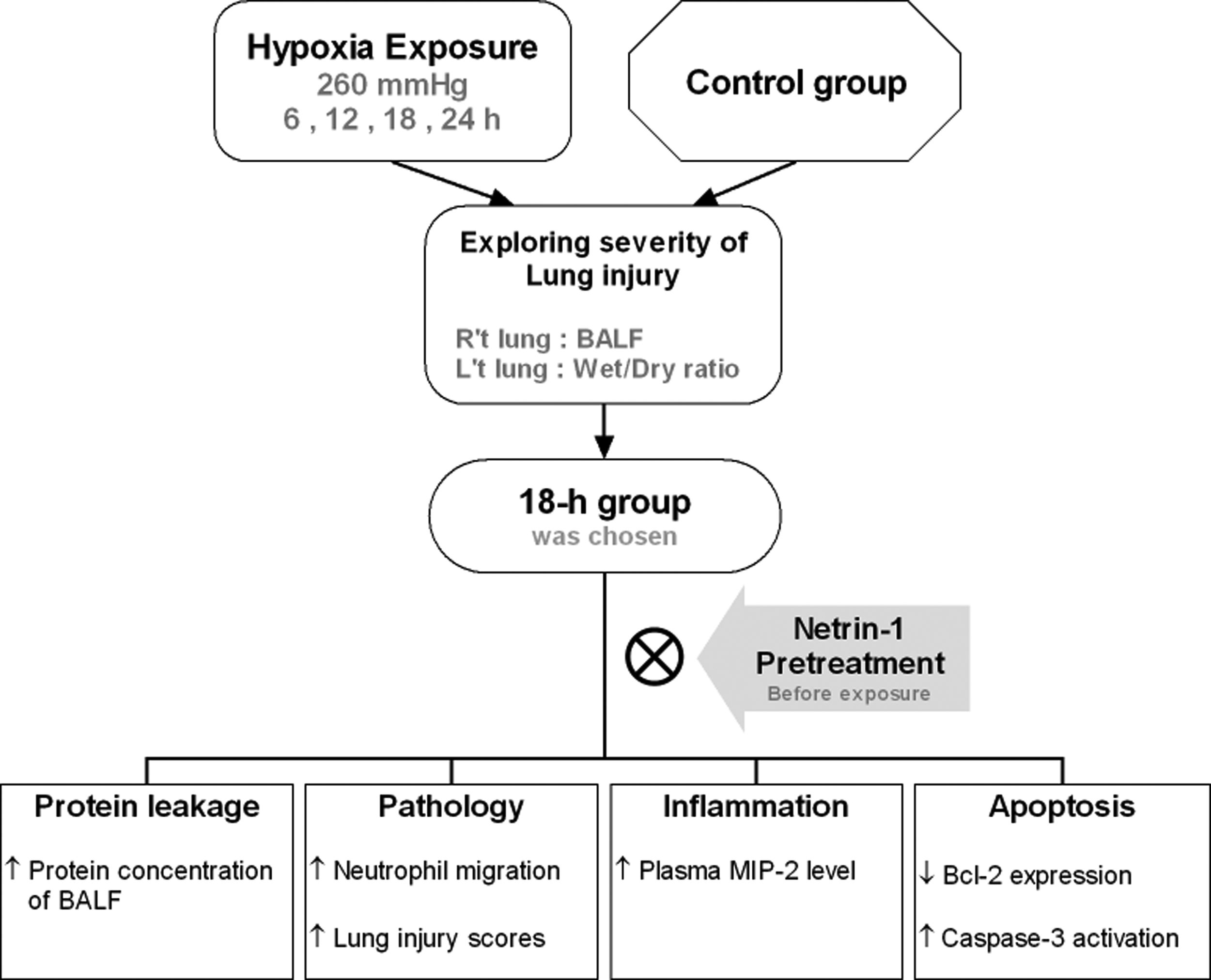
Flow diagram of the experimental protocol.
The adaption phase was necessary for the mice to reduce acute death rate in the initial 30 minutes due to high fluctuations in air pressure. The wet/dry weight ratio did not significantly differ among the groups. After exposure to 260 mmHg for 18 hours, leakage of protein was significantly severe compared with that in the control group; therefore, we chose 18 hours as the exposure period to investigate the effects of netrin-1.
The mechanism of high-altitude pulmonary edema (HAPE) consists of pulmonary vascular responses to hypoxia, which occurs in a heterogeneous pattern and imposes higher stresses in the overperfused areas (Schoene, 2008). This clinical condition was compatible with our pathological findings, where the injured parts of lungs were characterized by heterogeneous neutrophil infiltration and thickness of alveolar walls. A higher capillary density makes mice more resistant to hypoxia compared with human beings (Schmidt-Nielsen and Pennycuik, 1961), which might be the reason why pulmonary edema was not obvious in this study. The results of this study might represent subclinical HAPE.
During alveolar epithelial injury, macrophages secrete proinflammatory cytokines, including TNF-α, IL-6, and IL-1β, to trigger an inflammatory reaction (Tsushima et al., 2009; Grommes and Soehnlein, 2011). A previous study demonstrated that TNF-α level peaked at 1.5 hours and then quickly declined to the base level 12 hours after lipopolysaccharide (LPS) challenge in mice (Hong et al., 2009). The longer exposure period (18 hours) in our study made it difficult to detect TNF-α. Both damaged epithelial cells and macrophages secreted MIP-2 in mice to attract neutrophils (Ohtsuka et al., 2001). Neutrophil recruitment may also contribute to subsequent inflammatory response and tissue damage (Abraham, 2003; Grommes and Soehnlein, 2011). The elevated MIP-2 level and increased neutrophil counts might result from damaged epithelial cells after 18 hours of hypobaric exposure. Netrin-1 has been shown to inhibit leukocyte migration in vitro and in vivo and eliminate inflammation through netrin-1 receptor UNC5b (Ly et al., 2005). Furthermore, netrin-1 might also regulate the inflammatory response of neutrophils and macrophages by inhibiting COX-2-mediated PGE2 production (Ranganathan et al., 2013). Our results showed that neutrophil counts were significantly reduced, but the change in the thickness of septa was not prominent in the 18-hour exposure group pretreated with netrin-1 (data not shown). Therefore, lung injury score improved mainly due to decreased neutrophil infiltration.
Exposure to hypoxia impairs alveolar edema clearance by downregulating both epithelial sodium channel and the Na/K-ATPase function. Other adverse effects of hypoxia on alveolar cell function include increase in reactive oxygen species production and the triggering of apoptosis (Jain and Sznajder, 2005; Desireddi et al., 2010). Oxidative stress can cause cellular injury and increase vascular endothelial permeability (Lum and Roebuck, 2001). Protein leakage was noted in the 18-hour group, which improved upon netrin-1 pretreatment. Although there was no direct evidence to prove that netrin-1 could reduce oxidative stress, netrin-1 possesses the potential to eliminate further epithelial injury induced upon hypoxia to reduce protein leakage. Previous studies have indicated that netrin-1 promotes epithelial sodium channel-mediated alveolar fluid clearance via the activation of the adenosine 2b receptor in LPS-induced acute lung injury. Besides, the activation of the adenosine 2b receptor dampens hypoxia-induced vascular leakage (Eckle et al., 2008; He et al., 2014).
Oxidative stress triggers inflammation and/or apoptosis. In our study, inflammation might have caused lung injury initially, because of activated redox-sensitive transcription factor NF-κB and elevated plasma MIP-2 levels. Intrinsic pathway of apoptosis was also activated. The hypoxic insult resulted in changes in the inner mitochondrial membrane, including opening of the mitochondrial permeability transition pore, loss of mitochondrial transmembrane potential, and release of proapoptotic proteins. These proteins activate the caspase-dependent mitochondrial pathway. The activation of caspase-3 leads to the formation of apoptotic bodies through the execution pathway (Elmore, 2007). The members of the Bcl-2 family control and regulate these apoptotic mitochondrial events (Cory and Adams, 2002). Hypoxic stimuli activate the BH3-only proteins that could act as damage sensors. The BH3-only proteins inhibit the expression of Bcl-2 proteins to mediate the downstream apoptosis pathway (Adams and Cory, 2007; Lomonosova and Chinnadurai, 2008). Pretreatment with netrin-1 maintained homeostasis by reversing the changes in Bcl-2 expression induced upon hypobaric exposure. Furthermore, the prevention of apoptosis by netrin-1, as shown in our results, was consistent with other studies that investigated different pathways (Liao et al., 2013; Son et al., 2013; Chen et al., 2017). This might further support that netrin-1 could be beneficial in this condition.
The expression of netrin-1 increased slightly during 18-hour hypoxic exposure, which was comparable with Rosenberger's study. Hypoxic environment upregulates epithelial netrin-1 expression (Rosenberger et al., 2009). However, pretreatment with exogenous netrin-1 showed much more netrin-1 protein expression than hypoxic exposure in this study. A previous study showed that hypoxia could induce the downregulation of UNC5B receptor (Dakouane-Giudicelli et al., 2011). Our study presented similar results, and this condition could be reversed by pretreatment with exogenous netrin-1. As a result, netrin-1 might mediate part of its protective effect through UNC5HB receptor by inhibiting neutrophil migration (Ly et al., 2005).
The neuronal guidance molecule, netrin-1, plays a significant role in directed migration of leukocytes. In inflammatory bowel disease, leukocyte trafficking attracted by chemokines released from epithelium cells during infection or inflammation is a key feature of disease pathogenesis (Papadakis, 2004). Mice treated with exogenous netrin-1 were protected during the course of acute colitis by limiting neutrophil recruitment. The results were comparable with our study (Ly et al., 2005; Aherne et al., 2012). Mirakaj's murine study showed that pulmonary netrin-1 levels were suppressed during an acute lung injury induced by LPS. This resulted in an increased infiltration of neutrophils and severe pulmonary inflammation. Exogenous netrin-1 significantly reduced the degree of acute lung injury through the adenosine 2b receptor (Mirakaj et al., 2010). Aherne's study indicated that exogenous mouse netrin-1 could attenuate the severity of dextran sulfate sodium-induced colitis by suppressing neutrophil trafficking through the adenosine 2b receptor in mice (Aherne et al., 2012). Except for its protective effect, we should be cautious that netrin-1 could facilitate the formation of abdominal aortic aneurysms. Netrin-1 is a major signal that mediates the interaction between inflammation and chronic erosion of the extensive extracellular matrix in mice abdominal aortic aneurysms. Netrin-1 induces a robust intracellular calcium flux through its receptor neogenin-1 in controlling transcriptional regulation and persistent catalytic activation of matrix metalloproteinase-3 by vascular smooth muscle cells (Hadi et al., 2018).
Impaired endothelial barrier during hypoxia, sepsis, and inflammation causes increased vascular permeability, which results in fluid leakage and subsequent edema and organ dysfunction (Thompson et al., 2004). Hypoxia increases the extracellular ratio of ATP/ADP via lytic and nonlytic pathways (Bowser et al., 2018). Purinergic signaling events play an important role in controlling the vascular barrier function and concomitant inflammatory events. ATP receptor activation on vascular endothelial cells declines endothelial barrier function and thereby facilitates neutrophil–endothelial transmigration (Eltzschig et al., 2003). Meanwhile, neutrophil-dependent ATP released from the leading edge of a neutrophil is rapidly converted to adenosine by the CD39/CD73 pathway, which activates downstream adenosine receptor, particularly of endothelial adenosine 2b receptor. The adenosine 2a and 2b receptor signaling dampens inflammatory activation by resealing the endothelial barrier and restricting tissue infiltration of neutrophils (Sitkovsky et al., 2004; Eckle et al., 2008; Bowser et al., 2017). Hypoxia inducible factor (HIF)-1α–induced netrin-1 interacts with the adenosine 2b receptor on polymorphonuclear leukocytes, attenuating transepithelial migration and limiting tissue damage (Bowser et al., 2018). Previous studies have presented the protective role of adenosine signaling in injury to organs such as kidney, lung, heart, and intestines (Yuan et al., 2018). The adenosine 2b receptor, in particular, provides cardioprotective effects during myocardial ischemia/reperfusion injury (Eltzschig et al., 2013). Enhancing extracellular adenosine signaling or the targeting of individual adenosine receptors has shown promising effect in preventing or treating acute kidney injury from ischemia in murine models (Bauerle et al., 2011).
The interdependent relationships between hypoxia and inflammation are two sides of the same coin. On the one hand, an inflammatory disease causes tissue hypoxia. The shifts in metabolic supply and demand ratios due to increased oxygen demand and impaired oxygen provision lead inflamed tissues to become severely hypoxic. On the other hand, diseases primarily induced by hypoxia cause secondary inflammatory changes, such as subsequent inflammation after HAPE and ischemia/reperfusion injury following acute myocardial infarction (Bartels et al., 2013; Vohwinkel et al., 2015; Eltzschig and Carmeliet, 2011). Previous studies have shown that HIF-elicited gene programs dampened hypoxia-induced inflammation, which is implicated in lung protection through the enhanced production and signaling effects of anti-inflammatory signaling molecules, including adenosine and netrin-1 (Rosenberger et al., 2009; Eltzschig et al., 2012; Bartels et al., 2013).
There are some limitations in this study. Although severe lung edema was induced in a study by Tan et al. (2015), failure to set up a similar model could be explained by the differences in the use of mice species, protocol, and hypobaric chamber. Notwithstanding, the results of this study might represent conditions of subclinical HAPE. We did not measure the level of oxidative stress and vascular permeability because the major purpose was to establish an acute lung injury model induced upon hypobaric hypoxia in mice. Besides, the amount of blood sample and tissue extracted was not enough to examine all the parameters, including HIF-1α, which plays a critical role in the induction of hypoxia and the activation of NF-κB (Walmsley et al., 2005). However, Moon et al. (2016) indicated that hypobaric hypoxia (570 mmHg; exposure period 3 and 6 hours) upregulated serum mRNA levels of HIF-1α and VEGF in mice. The even more stringent experimental conditions used in our study might have also activated HIF-1α, but further investigations are needed.
Conclusion
We established a mouse model of acute lung injury induced by hypobaric hypoxia. Netrin-1 dampens lung injury by inhibiting the migration of neutrophils and attenuating apoptosis.
Footnotes
Acknowledgments
This study was supported by grants from the National Defense Medical Center, Taiwan, Wan Fang Hospital, Taiwan (107-wf-eva-33); Hualien Armed Forces General Hospital (805-C108-02); and Ministry of Science and Technology, Taiwan (MOST 106-2314-B-038-044).
Author Disclosure Statement
No conflicting financial interests exist.