
Abstract
Pu, Xiaoyan, Xue Lin, Xianglan Duan, Junjie Wang, Jun Shang, Haixia Yun, and Zhi Chen. Oxidative and endoplasmic reticulum stress responses to chronic high-altitude exposure during the development of high-altitude pulmonary hypertension. High Alt Med Biol. 21:378–387, 2020.
Objectives:
To investigate the effect of endoplasmic reticulum (ER) stress during the development of high-altitude pulmonary hypertension (HAPH) after chronic high-altitude exposure, as well as the association between oxidative stress and ER stress.
Methods:
Forty male Sprague-Dawley rats were placed in a low-pressure chamber with a simulated altitude of 4,200 m for 0–28 days. Rats were chosen at random on days 0, 7, 14, and 28 of chronic high-altitude exposure and were examined for pulmonary arterial pressure, oxidative stress, apoptosis, and ER stress.
Results:
Chronic high-altitude exposure caused a continuous deterioration of pulmonary hypertension, which was accompanied by obvious apoptosis of alveolar epithelial cells and remodeling of pulmonary vessels. From day 7 of high-altitude exposure, although the activities of glutathione peroxidase and superoxide dismutase were gradually decreased, the generation of both malondialdehyde and reactive oxygen species was increased in a time-dependent manner. The protein expression of ER stress-related GRP78, PERK, IRE1α, ATF6, ATF4, CHOP, and caspase-12 in lung tissue was significantly upregulated from day 14 of high-altitude exposure. Further, the expression of caspase-12 in alveolar epithelial cells and vascular smooth muscle cells was also increased from day 14 of high-altitude exposure.
Conclusions:
Early high-altitude exposure first activates oxidative stress; then, it gradually activates ER stress. The activation of ER stress might promote the apoptosis of alveolar epithelial cells and the remodeling of pulmonary vessels by exacerbating the oxidative stress response during the development of HAPH after chronic high-altitude exposure.
Introduction
As an outcome of chronic alveolar hypoxia and a feature of congestive right heart failure and high-altitude pulmonary edema in subacute mountain sickness and chronic mountain sickness of the final stage of chronic mountain sickness in the Himalayas and Andes (Maggiorini and Leon-Velarde, 2003), high-altitude pulmonary hypertension (HAPH) results in hypoxic vasoconstriction and remodeling of the pulmonary circulation (West, 2013). The physiological reaction of the pulmonary circulation to normobaric and hypobaric hypoxia is to enhance pulmonary arteriolar resistance.
However, chronic exposure to high altitude will cause persistent pulmonary hypertension among rats because of hypoxic vasoconstriction, with related reconstruction of pulmonary arteries and right ventricular hypertrophy (Omura et al., 2000; Swenson et al., 2002). The pathogenesis of HAPH is not fully understood, but hypoxic stimuli inducing the alteration of ion channel activity, oxidative stress injury, and inflammatory cytokines have been implicated in the subsequent increase in pulmonary arterial vasoconstriction and vascular remodeling (Mandegar et al., 2004; Nozik-Grayck and Stenmark, 2007).
As a major disturbance, oxidative stress from hypoxia causes endoplasmic reticulum (ER) dysfunction (Amodio et al., 2018). Unfolded and/or misfolded proteins in the ER are accumulated by ER stress, and according to the latest research, ER stress is involved in various diseases such as inflammatory diseases and proliferative diseases (Marciniak and Ron, 2006; Pluquet et al., 2015). Newly synthesized protein moves to the ER lumen, where protein molecules fold into appropriate three-dimensional structures by means of chaperone proteins and the proper levels of calcium and adenosine triphosphate. Nevertheless, a lot of noxious extracellular stimuli cause the disorder protein-folding processes and result in ER stress, which is embodied as the unfolded protein response (UPR) in cells (Marciniak, 2017).
Three branches of signaling triggered by PKR-like ER kinase (PERK), inositol-requiring enzyme-1 (IRE1), and activating transcription factor-6 (ATF6) in the ER membrane (Breckenridge et al., 2003) are activated by the UPR. The initiation of ER stress depends on the UPR, which promotes activation of transmembrane proteins PERK, IRE1, and ATF6 by competitive binding with Bip/GRP78, followed by the dissociation of Bip/GRP78, which upregulates the proapoptosis transcription factor CHOP to make active the ER stress-mediated apoptosis pathway (Nakagawa et al., 2000; Dong et al., 2009).
Although unique apoptotic pathways can be triggered by the ER, a reaction to different stress signals, it is proved that the ER can also collaborate with other apoptotic pathways (Lemasters, 2005). According to current accumulated evidence, oxidative stress aggrandizes ER stress in stress-damaged cells and vice versa (Tan and Chiu, 2013). There have been several reports showing that oxidative stress-induced apoptosis occurred with remodeled pulmonary vasculature after acute or chronic high-altitude exposure (Jefferson et al., 2004; Xu et al., 2015). Oxidative stress or hypoxia can induce ER stress to promote apoptosis; nevertheless, it is unclear whether ER stress participates during the development of HAPH.
Therefore, to further explore the pathogenesis of HAPH, we investigated the ER stress in the development of HAPH in Sprague-Dawley (SD) rats after chronic high-altitude exposure, as well as the relationship of oxidative stress to ER stress.
Materials and Methods
Animals
The Institutional Animal Care and Use Committee of Qinghai University approved all experimental protocols and methods pertaining to animals. All experiments used 40 male SD rats of 250–300 g. Rats were exposed to a low-pressure chamber with a simulated altitude of 4,200 m for 20 hours per day ranging from 0 to 28 days. The barometric pressure was lowered stepwise to reach an altitude corresponding to 4,200 m (atmospheric pressure = 450.42 mmHg, PO2 = 12.579 kPa) after six exposures. Rats were categorized into four groups (10 per group) based on high-altitude exposure duration: 0, 7, 14, or 28 days. The animals were sacrificed the day after the final hypoxic exposure. During hypoxic exposure, all rats were housed and caged every five rats with a 12:12-hour light–dark cycle and supplied with food in an unrestricted way.
Measurements of key physiological indicators
After each exposure time, after taking out rats from the low-pressure chambers and placing them under a plastic hood for 10–15 minutes before sacrifice, a gas mixture consisting of 10.5% O2 in N2 at sea level pressure was used to flush them. With the equivalent partial pressure of arterial oxygen (PaO2) as in the low-pressure chamber, this gas mixture ensures that the animals exposed to altitude remained hypoxic whey they died. An intraperitoneal injection of 15 mg/kg xylazine (Bayer, Germany) and 90 mg/kg S-ketamine (Pfizer, Ireland) anesthetized and heparinized (250 U) all rats.
For normal body temperature, rats were put on a heating plate. For mechanical ventilation with a tidal volume of 6 ml/kg at 80 per minute and a positive end-expiratory pressure (PEEP) of 2 cm H2O and an inspiration-to-expiration ratio of 1:1.5 (Hugo Sachs Electronics, March-Hugstetten, Germany), a tracheal tube was inserted. To prevent atelectasis, the PEEP was increased from 3 to 10 cm H2O for 10 seconds every 15 minutes.
After inserting a catheter filled with saline into the jugular vein, right ventricular systolic pressure (RVsP) was recorded as a surrogate for systolic pulmonary artery pressure (PAP) by advancing it into the right ventricle. Pressure curves were used to monitor proper catheter placement. Systemic systolic blood pressure (FAsP) was recorded by inserting another saline-filled catheter into the femoral artery. After connecting pressure transducers with amplifiers (DBA; Hugo Sachs Electronics), PowerLab (AdInstruments, Speckbach, Germany) was used to digitize the output signal, which was recorded continuously on a PC. Systolic values from pressure curves and heart rate were obtained by detecting pressure peaks and frequency with the PowerLab software, respectively.
After measurements of PAP, PaO2, and arterial oxygen saturation (SaO2), the heart with ice-cold phosphate buffer saline was used to perfuse all rats under anesthesia. After perfusion, the middle left lung lobe of five rats per group was removed and fixed in 4% paraformaldehyde for histological analyses; the left lung tissues of another five rats were stored at −80°C until analysis.
Biochemical reaction
Ice-cold Tris-HCl buffer (pH 7.4, 50 mM) and Ultra-Turrax T 25 Basic Homogenizer (IKA-Werke GmbH & Co., Staufen, Germany) at 4°C were applied to prepare the homogenate of lung tissues. The supernatant was separated from the homogenate after a 15-minute centrifugation at 800 g for further application in various biochemical researches. The thiobarbituric acid method was applied to detect the bioactivity of malondialdehyde (MDA); the xanthine oxidase method was adopted to determine the activity of superoxide dismutase (SOD); and the dithiodinitrobenzoic acid method was employed to measure the activity of non-enzymatic antioxidant glutathione peroxidase (GSH-Px) with the corresponding commercial biochemical kits (Nanjing Built Biology, Nanjing, China) on basis of the manufacturer's instructions.
Reactive oxygen species detection in lung tissues
The level of reactive oxygen species (ROS) generation was decided as described by Yang et al. (2015). Briefly, after mixing 50 μL of freshly prepared homogenate of lung tissues with 4.85 ml potassium phosphate buffer (pH 7.4, 100 mmol/L), a 15-minute incubation of the mixture was made with 2′, 7′-dichlorofluorescin diacetate (DCFH-DA) in a final concentration of 5 μmol/L methanol at 37°C. A ten-minute centrifugation of dye-loaded samples was made at 4°C and 12,500 g. After mixing the pellet on a vortex at 0°C in 5 ml of 100 mmol/L phosphate buffer (pH 7.4), a 6-minute incubation of mixture was made again at 37°C. Fluorescence spectrophotometry (AquaMate 8000 UV-Vis Spectrophotometer) was used to measure fluorescence at wavelengths of 525 nm for emission and 488 nm for excitation. The cuvette holder was kept at 37°C. A dichlorofluorescin standard curve in methanol was used to quantify the levels of ROS.
Histopathological analyses of hematoxylin and eosin and Sirius-red staining
After 24 hours of paraformaldehyde fixation, the middle left lung lobe was embedded in paraffin and sectioned at 5 μm intervals. The lung slices were dewaxed, hydrated, and finally processed by hematoxylin and eosin and Sirius-red staining, before assessing histopathology. A Nikon digital imaging system (Nikon Eclipse 80i, Nikon, Japan) was used to capture images.
Two slices per animal and five animals per group were used to select 16 non-overlapping fields from each slide. The point-counting method of using a 10 × 10 grid on each image to count the ratio of intersections falling on air space to intersections on tissue at × 100 magnification was used to determine the ratio of air space to tissue. Pulmonary arterioles of 50–100 μm in diameter were selected to calculate the medial layer thickness index (medial layer thickness/vascular wall thickness) and the medial layer area index (medial layer area/vascular wall area) at × 400 magnification by Image-Pro 6.0 software (Media Cybernetics, Silver Spring, MD).
Apoptosis analysis in lung tissues
The terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) method was used to detect apoptosis by using the DeadEnd Colorimetric Apoptosis Detection system (Promega, MDAison, Wisconsin). After digesting proteinase and removing endogenous peroxidase, a mixture containing fluorescein isothiocyanate-labeled dUTP and TdT was used to incubate the sections. The peroxidase labeled with anti-fluorescein isothiocyanate antibody was used to treat the sections. After being developed with 3,3[prime]-diaminobenzidine tetrahydrochloride, the reaction products were counterstained with methyl green. The counting of positive cells for TUNEL was made in 20 randomly selected fields per section under a microscope with × 250 magnification. The percent of epithelial cells positive was employed to present the number of positive cells.
Immunohistochemistry
Briefly, Anti-rat caspase-12 antibody (1:1,000; Boster Biotechnology, Wuhan, China) and appropriate secondary antibody (Boster Biotechnology) at 37°C were used to incubate lung tissue sections overnight at 4°C. After 30 minutes, peroxidase-conjugated avidin and diaminobenzidine were used to visualize bound antibodies based on the manufacturer's instructions (Boster Biotechnology). ImageJ program image analysis software (Image J2X; Media Cybernetics, Carlsbad, CA) was adopted to quantify and average staining signals.
Western blotting
A polytron homogenizer (Kinematica, Luzern, Switzerland) was used to homogenize frozen lung tissues in buffer A containing 5 mM MgCl2, 25 mM Hepes (pH 7.5), 1 mM EGTA, 1 μg/ml leupeptin, 1 mM phenylmethyl sulfonyl fluoride, and 1 μg/ml aprotinin. After a 10-minute incubation on ice, extracts were vortexed for 10 seconds and centrifuged into pellet nuclei at 800 g for 2 minutes at 4°C. Thirty-minutes centrifugation of supernatant was made at 15,000 g and 4°C. Western blot analysis with extracted protein was made for caspases except for cleaved caspase-3.
After extraction and quantification, 5-minute boiling in sodium dodecyl sulfate (SDS) sample buffer was used to denature the protein supernatant. After being separated by 6%–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), 40 μg of total protein was blotted onto polyvinylidene difluoride membranes, and then probed with the following antibodies: rabbit anti-caspase-3 (1:1,000; Abcam), rabbit anti-cleaved caspase-3 (1:1,000; Abcam), rabbit anti-GRP78 (1:1,000; Abcam), rabbit anti-PERK (1:1,000; Abcam), rabbit anti-Phospho-PERK (1:1,000; Thermo) rabbit anti-ATF6 (1:1,000; Abcam), rabbit anti-ATF4 (1:1,000; Abcam), rabbit anti-IRE1α (1:1,000; Abcam), rabbit anti-Phospho-IRE1α (1:1,000; Abcam), rabbit anti-CHOP (1:1,000; Abcam), rabbit anti-caspase-12 (1:1,000; Abcam), and anti-β-action (1:1,500; Sigma) and conjugated to secondary antibodies of horseradish peroxidase.
After being visualized by a 1-minute incubation with BeyoECL Plus (P0018, Beyotime, China), protein bands were imaged by a Gel Image System (Tanon, 5200, China). Image J software was used to perform densitometry.
Statistical analysis
Analyses were conducted with SPSS 20.0 software. The mean ± standard deviation was used to express data. The levels of significance were calculated by using two-tailed Student's t-tests and post hoc analysis of variance. A p-value <0.05 was considered significant.
Results
Effects of chronic high-altitude exposure on pulmonary hypertension and oxidative stress
With the increase in PAP over the 4-week experimental period in rats exposed to a simulated 4,200 m asl, a significant increase in PAP was observed on the 7th and 14th day after high-altitude exposure, as shown in Figure 1A. Both the PaO2 and SaO2 showed a notable tendency to fall (Fig. 1B, C), suggesting that the pulmonary hypertension induced by hypoxia progressed with prolongation of chronic high-altitude exposure. It was shown that the rats increasingly and obviously lack physical activity and appetite with the prolonged high-altitude exposure, which stands for heart failure comparable to cachexia in patients with HAPH.

Effects of high-altitude exposure on pulmonary hypertension and oxidative stress.
The level of ROS generation was measured spectrofluorimetrically (Fig. 1D), and ROS generation in lung tissues was significantly increased with the prolongation of high-altitude exposure (Fig. 1E). Levels of the ROS scavengers SOD and GSH-Px in lung tissue were significantly reduced in a time-dependent manner with high-altitude exposure (p < 0.001) (Fig. 1F, G). The level of MDA in lung tissue showed an increased tendency after chronic high-altitude exposure, and the rats were significantly different from those in the chronic high-altitude exposure group (Fig. 1H) in the control group (0 days) at day 14. According to these data, a progressive oxidative stress injury in lung tissue was caused by chronic high-altitude exposure.
Effects of chronic high-altitude exposure on lung structure and apoptosis
By comparing with the control group (0 days), the hypoxia caused by chronic high-altitude exposure widened alveolar septa as the corresponding number of alveoli and thickened the tunica media of pulmonary arterioles with collagen fiber proliferation reduced apparently, which worsened over the duration of chronic high-altitude exposure (Fig. 2A). Further, the ratio of air space to tissue decreased gradually with altitude exposure (Fig. 2B), whereas the medial layer thickness index and area index showed a notable tendency to augment (Fig. 2C, D), suggesting that hypoxia-induced pulmonary hypertension was associated with pulmonary vascular remodeling.
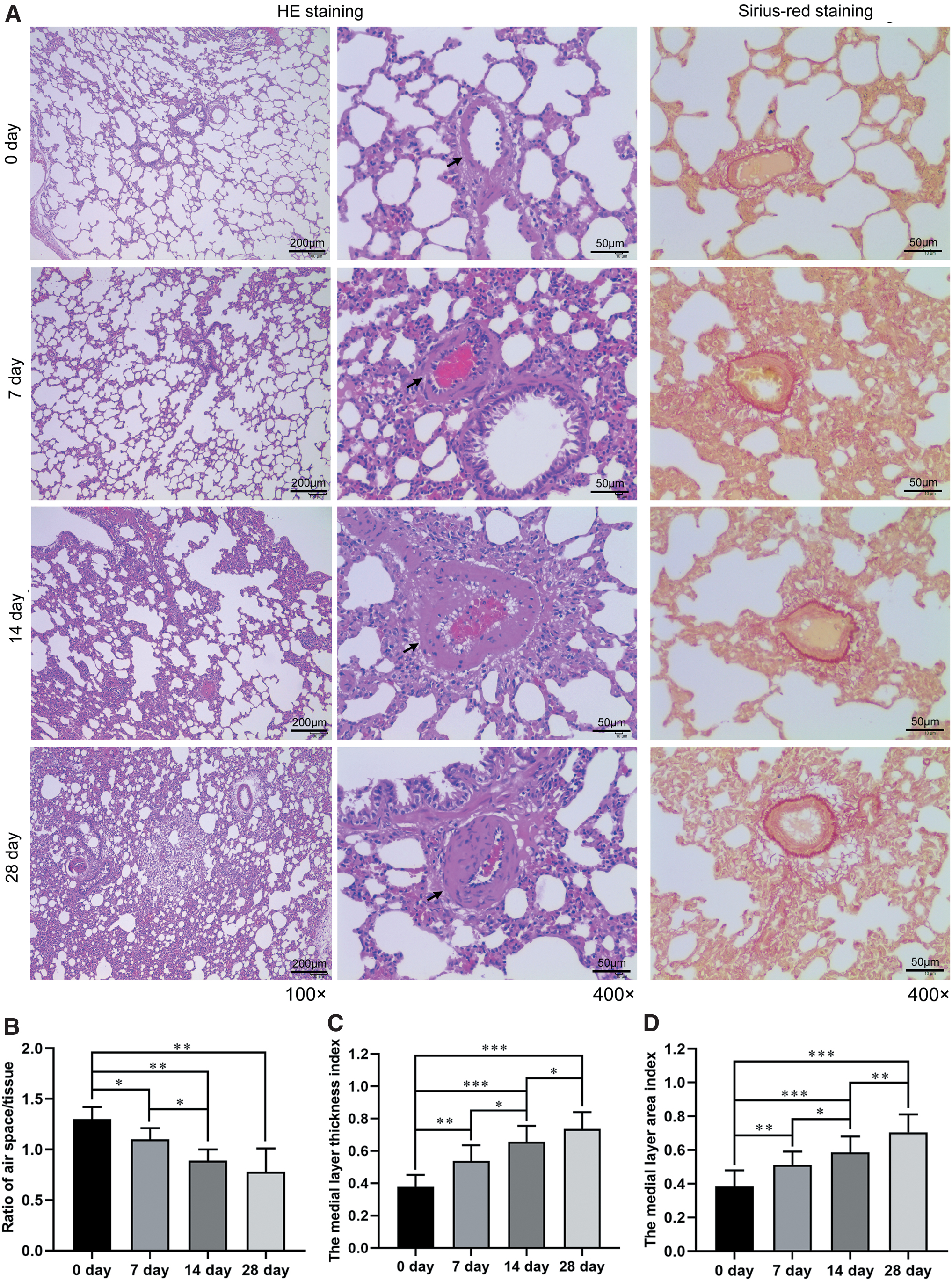
Effects of high-altitude exposure on lung structure and pulmonary vascular remodeling.
Positive signals for TUNEL were mainly detected in the nuclei of alveolar epithelial cells of lung tissues (Fig. 3A). The TUNEL-positive cells for apoptosis rate of alveolar epithelial cells were greatly increased after day 14 and 28 of high-altitude exposure, compared with day 0, whereas no difference was observed between day 0 and 7 after high-altitude exposure (p < 0.001) (Fig. 3B). Further, the expression of caspase-3 and the active form of caspase-3 (cleaved caspase-3), as well as the ratio of cleaved caspase-3/caspase-3, were significantly upregulated in day 14 and 28 after high-altitude exposure, but no difference was observed between day 0 and 7 (Fig. 3C, D). Those data suggested that the apoptosis of alveolar epithelial cells did not occur in the early stage of HAPH (0–7 days) but increased after chronic high-altitude exposure (14–28 days).

Effects of high-altitude exposure on apoptosis of alveolar epithelial cells.
ER stress response in lung tissue after chronic high-altitude exposure
As displayed in Figure 4A and B, the expression of ER signaling proteins, such as GRP78, PERK, p-PERK, IRE1α, p-IRE1α, ATF6, ATF4, and CHOP, as well as the ER stress-medicated apoptosis protein caspase-12, were significantly upregulated on the 14th and 28th day after high-altitude exposure. Moreover, the increased ratios of p-PERK/PERK and p-IRE1α/IRE1α further confirmed that hypoxia induced the activation of ER signaling on day 14 and 28 after high-altitude exposure (Fig. 4C).
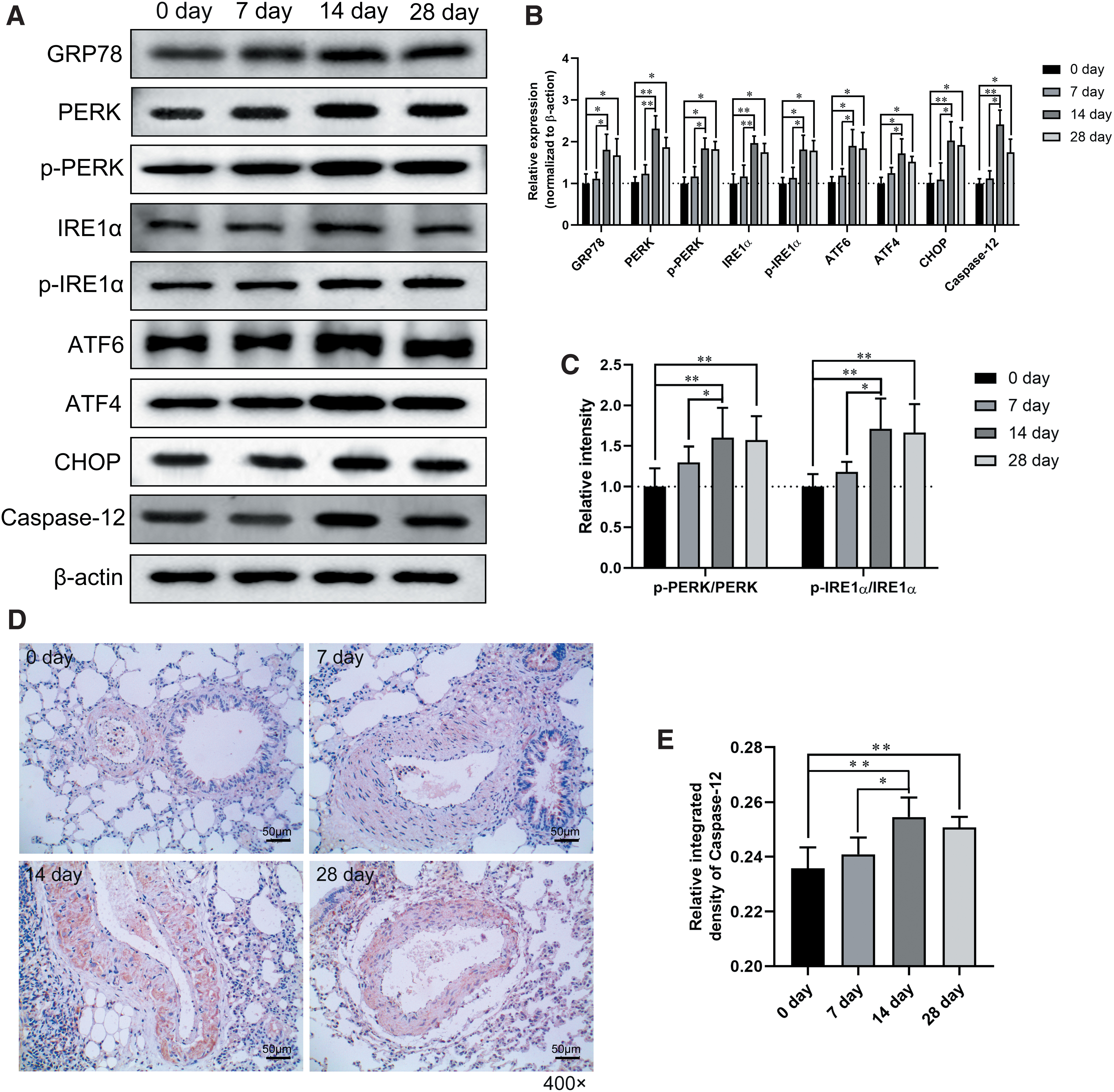
Endoplasmic reticulum stress response in lung tissue after high-altitude exposure.
Nevertheless, ER stress-related protein expression was not different between day 0 and 7 in the development of HAPH after chronic high-altitude exposure. A positive signal for caspase-12 was predominantly detected in the alveolar epithelial cells and vascular smooth muscle cells (Fig. 4D), and the relative integrated density of caspase-12 in lung tissues was greatly increased after day 14 and 28 of high-altitude exposure (Fig. 4E). These data suggested that the ER stress response was activated in chronic high-altitude exposure (14–28 days) but not the early stage of HAPH (0–7 days).
Discussion
In this article, we demonstrated the presence of oxidative stress and ER stress during the development of HAPH after increasing the duration of chronic high-altitude exposure. Both ER stress and oxidative stress were activated in a time-dependent manner and gradually deteriorated after chronic high-altitude exposure. The activation of ER stress promoted apoptosis of alveolar epithelial cells, which might be one of the predisposing causes of HAPH.
Hypoxia-induced oxidative stress exerts a crucial effect on the development of pulmonary hypertension after high-altitude exposure (Jaitovich and Jourd'heuil, 2017). Whether activated oxidative stress is a minor factor or a main contributor in the serious altitude illness is unknown, but certainly, exercise during the early hours of altitude exposure leads to an obvious increase in the incidence and severity of acute mountain sickness (Roach et al., 2000).
Indeed, the majority of people can bear and even stand mild oxidative stress and often make a response by synthesizing extra antioxidant defenses; however, serious oxidative stress can prevail over antioxidant defenses and trigger the generation of ROS, which may result in impaired muscle function and decreased capillary perfusion at altitude or may even exert an effect on the precipitation of more serious neurological and pulmonary crises (Askew, 2002). A high level of oxidative stress causes an excessive increase in intracellular free calcium, destruction of energy metabolism, and damage to cellular organelles, such as proteins, lipids, and DNA (Rahal et al., 2014), so as to make a contribution to it.
Further, chronic hypoxia induces mitochondrial dysfunction by altering the redox potential of cells, which form superoxide (Yamamoto et al., 2006) by the direct single-electron reduction of molecular oxygen. The products of oxidative damage and activities of antioxidant enzymes were measured only by indirectly assessing oxidative stress. GSH-Px and SOD are central agents in cellular antioxidant defense; thus, changes in activity have been used to identify oxidative stress in previous studies (Rahal et al., 2014). SOD and GSH-Px are important scavengers of ROS. If ROS scavenger activities decrease, the number of free radicals would exaggeratedly increase, which is harmful to lipids, proteins, and nucleic acids.
As a highly toxic molecule, MDA is the main product of polyunsaturated fatty acid peroxidation; it is considered a marker of lipid peroxidation (Del Rio et al., 2005), and thus it is a product of oxidative damage. The interaction of MDA with proteins or DNA has been used to describe as potentially atherogenic and mutagenic (Del Rio et al., 2005). We found that activity of the antioxidant enzymes SOD and GSH-Px was decreased in a time-dependent manner after chronic high-altitude exposure. Further, MDA levels were increased. Therefore, hypoxia could induce oxidative stress and lipid peroxidation through the reducing activity of antioxidant enzymes.
Hypoxia is the main cause of pulmonary hypertension in chronic high-altitude exposure, and accordingly, we observed a time-dependent increase in PAP and reduction of PaO2 and SaO2 after chronic high-altitude exposure, which indicated that hypoxia occurred in the process of HAPH. Further, hypoxia generates oxidative stress, which has previously been implicated in pulmonary vascular remodeling (Matsui et al., 2004; Astorga et al., 2018). In our study, both hypoxia and oxidative stress occurred progressively in HAPH, and the apoptosis of alveolar epithelial cells was also increased after chronic high-altitude exposure. Apoptosis depends on the cascade signal transduction of caspase protein, which promotes cleaving caspase-3 into its active form, thus triggering a caspase signaling cascade of apoptosis (Chen et al., 1998).
It is observed that the expression of caspase-3 and the cleaved, active form of caspase-3 were significantly upregulated on day 14 and 28 after high-altitude exposure, which conformed to the occurrence of apoptosis. Further, pulmonary vascular remodeling was accompanied by apoptosis of alveolar epithelial cells and aggravated by the prolongation of high-altitude exposure. Some studies have suggested that chronic hypoxemia can lead to a constant increase in pulmonary vascular resistance and smooth muscle cell proliferation, resulting in pulmonary hypertension (Veres Racamonde et al., 2002; Vodoz et al., 2009; Porteous and Fritz, 2014).
Thus, we assumed that the destruction of pulmonary capillaries and vessels induced by apoptosis might cause an increase in pulmonary vascular resistance and PAP, thereby aggravating the effects of hypoxia, potentiating pulmonary remodeling, and eventually leading to pulmonary hypertension. However, the apoptosis of alveolar epithelial cells was not obvious in the early stages of chronic high-altitude exposure but was significantly activated from the 14th day after chronic high-altitude exposure.
Hypoxia or loss-of-function mutations can cause ER stress and thus activate the UPR; accordingly, ER stress-mediated apoptosis means a newly discovered apoptosis pathway (Beretta et al., 2010). Under normal circumstances, IRE1α, PERK, and ATF4/6 bind to the Bip/GRP78 complex and maintain an inactive state. Under stress, the UPR promotes the activation of the three transmembrane proteins by competitive combining with the Bip/GRP78 (Dong et al., 2009). The rapid upregulation of GRP78 marks the beginning of ER stress (Xia et al., 2017). CHOP is a significant pro-apoptosis transcription factor induced by ER stress, which is upregulated by the dissociated IRE1α, PERK, and ATF6 (Gwak et al., 2016; Pinto et al., 2016; Simard et al., 2016). CHOP, as a transcription element, can inhibit the expression of Bcl-2 and thus weaken its anti-apoptosis properties (Li et al., 2017).
Further, upregulated caspase-12, which is specifically expressed in the ER, also represents the activation of apoptosis via the ER pathway (Nakagawa et al., 2000). Wu et al. (2016) reported that pulmonary hypertension induced by monocrotaline was accompanied by ER stress-related gene activation, such as PERK, ATF6, and GRP78, suggesting that ER stress is activated during pulmonary hypertension. In our study, the upregulated expression of ER stress marker protein GRP78, and ER stress-related PERK, ATF6, ATF4, and CHOP were observed in the lung tissue of rats after chronic high-altitude exposure. Further, the increased expression of caspase-12 also indicates the activation of the apoptosis pathway caused by ER stress. Therefore, the occurrence of ER stress might aggravate apoptosis in the development of HAPH.
In our study, we found no difference in the expression of proteins related to ER stress between day 0 and 7 of high-altitude exposure, whereas ER stress was activated from day 14 after high-altitude exposure. Interestingly, we found that the change in the oxidative stress-related enzyme activity occurred earlier than the activation of ER stress. According to the accumulated lines of evidence, related to the generation of ROS, ER stress could induce apoptosis (Tan and Chiu, 2013).
Oxidative stress is triggered by chronic hypoxemia as ROS produce, which alters the reactions depending on cellular redox and interferes with protein-folding ability, which includes protein disulfide bonding; protein misfolding in the ER (Malhotra and Kaufman, 2007) is caused ultimately. Paradoxically, intracellular ROS production is increased by ER stress; ROS production in the ER lumen is enhanced by the increased protein disulfide bonding, and cytosolic Ca2+ is increased by the alteration of ER Ca2+ homeostasis, which stimulates mitochondrial ROS production (Zinszner et al., 1998; Yokouchi et al., 2008). Thus, we speculated that early exposure to high altitude induces oxidative stress and might promote the activation of ER stress in the development of HAPH, whereas the activation of ER stress may, in turn, contribute to ROS production.
Moreover, we found that neither the apoptosis level in alveolar epithelial cells nor ER stress appeared to continuously rise from day 14 to 28, which appears to be associated with a self-protection mechanism after long-term hypoxia. Some studies have demonstrated that the double-stranded RNA-activated PERK pathway of UPR attenuates the accumulation of intracellular ROS induced by ER stress, which simultaneously activates oxidative stress-inducible transcription factor Nrf2 to maintain the redox homeostasis, thereby ensuring cell survival (Cullinan and Diehl, 2004; Back et al., 2009; Ma et al., 2018). According to these researches, ROS generation sensitizes cells to ER stress, thereby contributing to apoptosis, but the increased oxidative stress burden associated with UPR activation can be buffered by PERK-dependent signaling through the promotion of Nrf2 activation.
However, the mechanisms by which ER stress-mediated PERK signaling interacts with Nrf2 regulation of deteriorated oxidative stress needs further study in the development of HAPH.
In conclusion, we found that oxidative stress and ER stress would be activated successively after chronic, simulated high-altitude exposure to 4,200 m asl, similar to living on a high-altitude plateau. The activation of ER stress might promote the apoptosis of alveolar epithelial cells and the remodeling of pulmonary vessels by exacerbating the oxidative stress response in the development of HAPH after chronic high-altitude exposure.
Footnotes
Acknowledgments
Authors' Contributions
Conceived and designed the experiments: X.P. and Z.C.; Performed the experiments: X.L., X.D. and H.Y.; Analyzed the data: J.W. and J.S.; Contributed reagents and materials: Z.C.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The Second Tibetan Plateau Scientific Expedition and Research Program (STEP), Grant No. 2019QZKK0606 supported this research; Project 31660254 of the Chinese National Nature Science Foundation; Project 2017-ZJ-Y13 of the Key Laboratory of Medicinal Animal and Plant Resources of the Qinghai-Tibetan Plateau; Project 2019-ZJ-7042 of the Department of Science and Technology of Qinghai Province; and the Young People of the Medical College of Qinghai University, Project 201910743003 of the National Students' Platform for Innovation and Entrepreneurship Training Program.